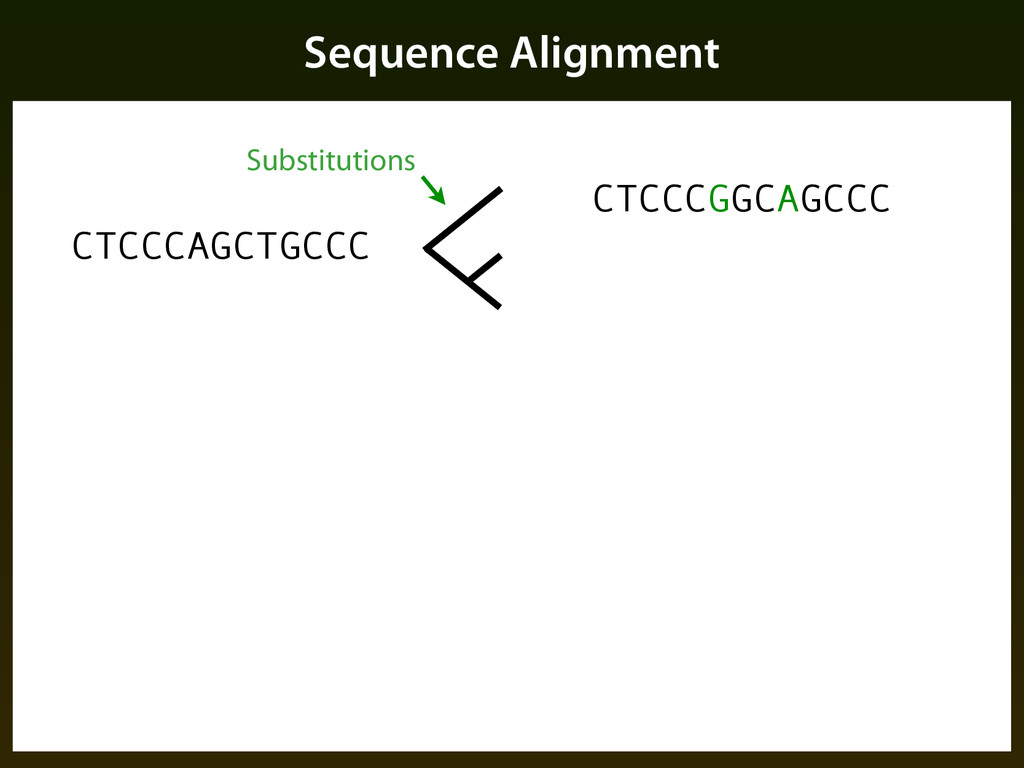
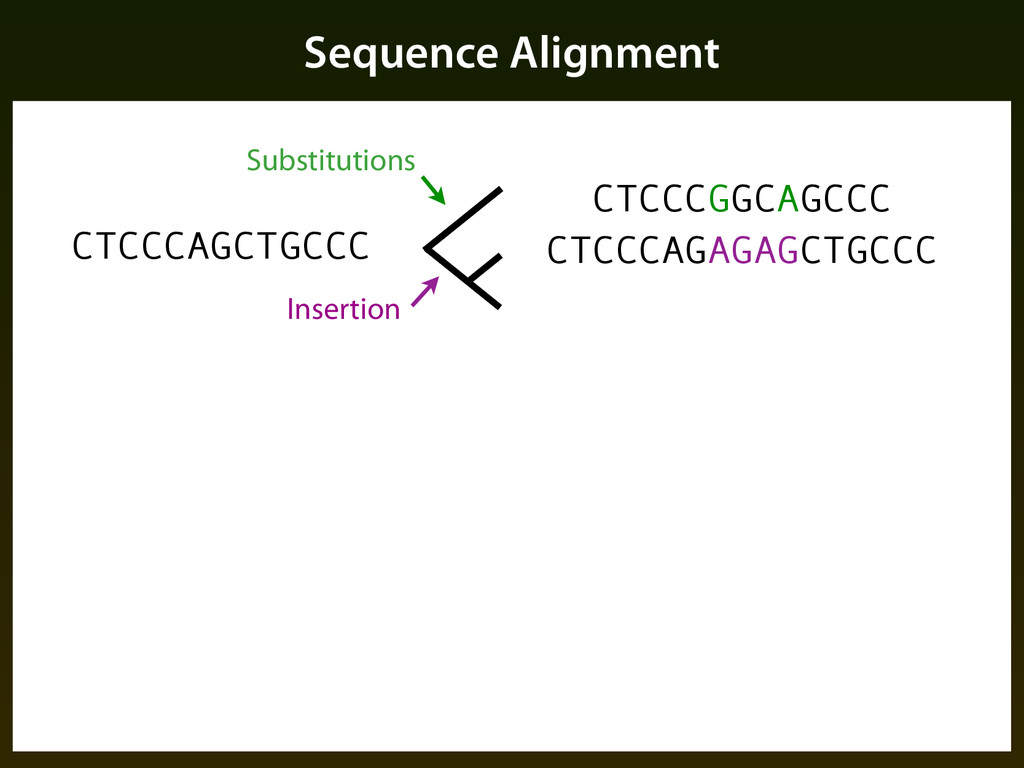
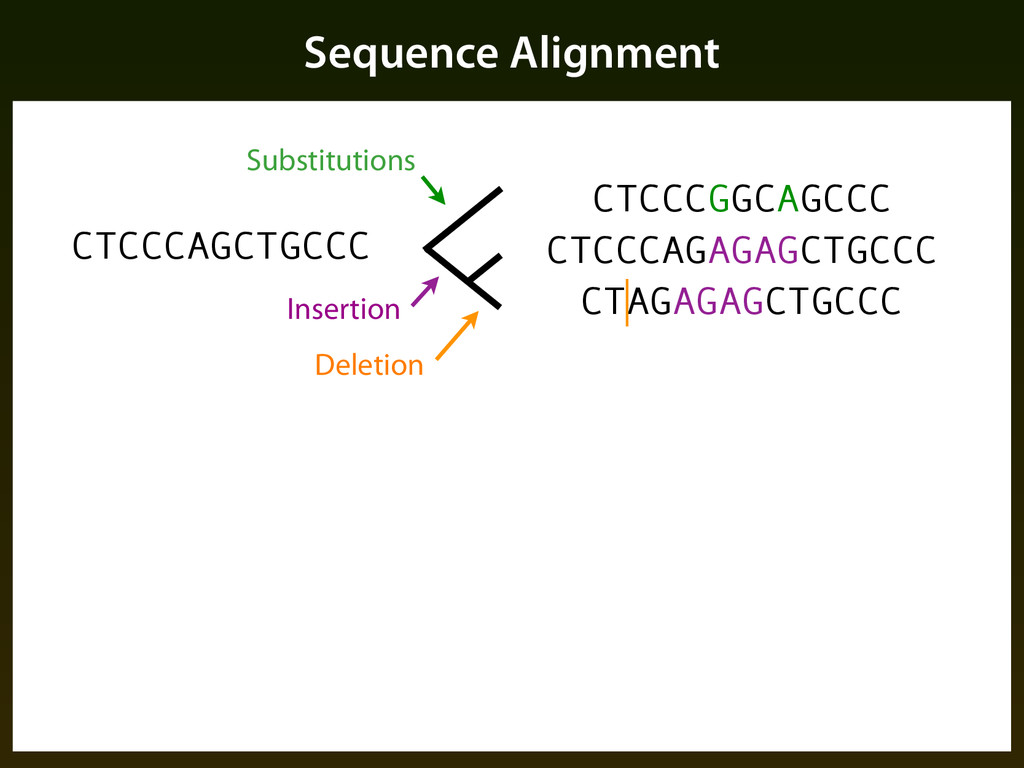
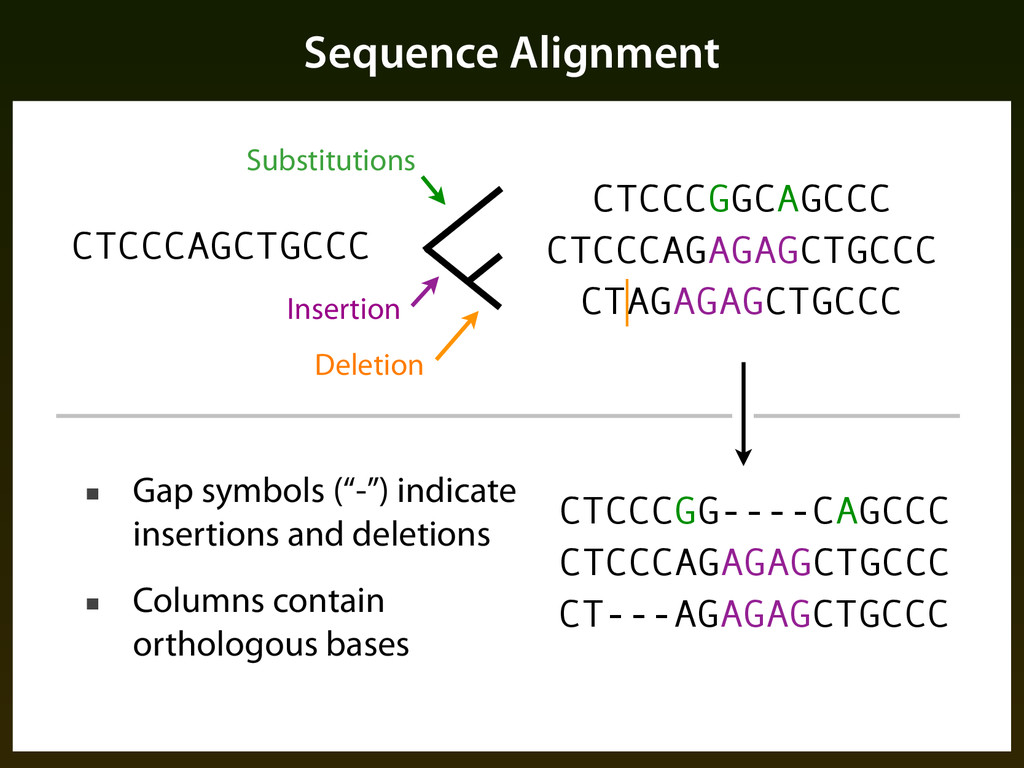
sequence patterns (“motifs”) ▪ Base composition ▪ Evolutionary constraint: ▪ Mutation events occur randomly, but selection determines if events are tolerated, resulting in a different pattern of change in functional regions
smaller alphabet that maintains the “right” information for some classification problem CTCCCAGCTGCCCAGTGCCGCCTCTTTTT CTCCTAGCTG-CCAGCATCTCCCGTTTTT CTCCCAGCTGCCCTGCGCCTCCTCTTTTT ↓ 13111021321110232112113133333
new agglomerative approach (allows us to scale to many more species) ▪ Improved handling of missing data (now used for all applications) ▪ It finally has a name!
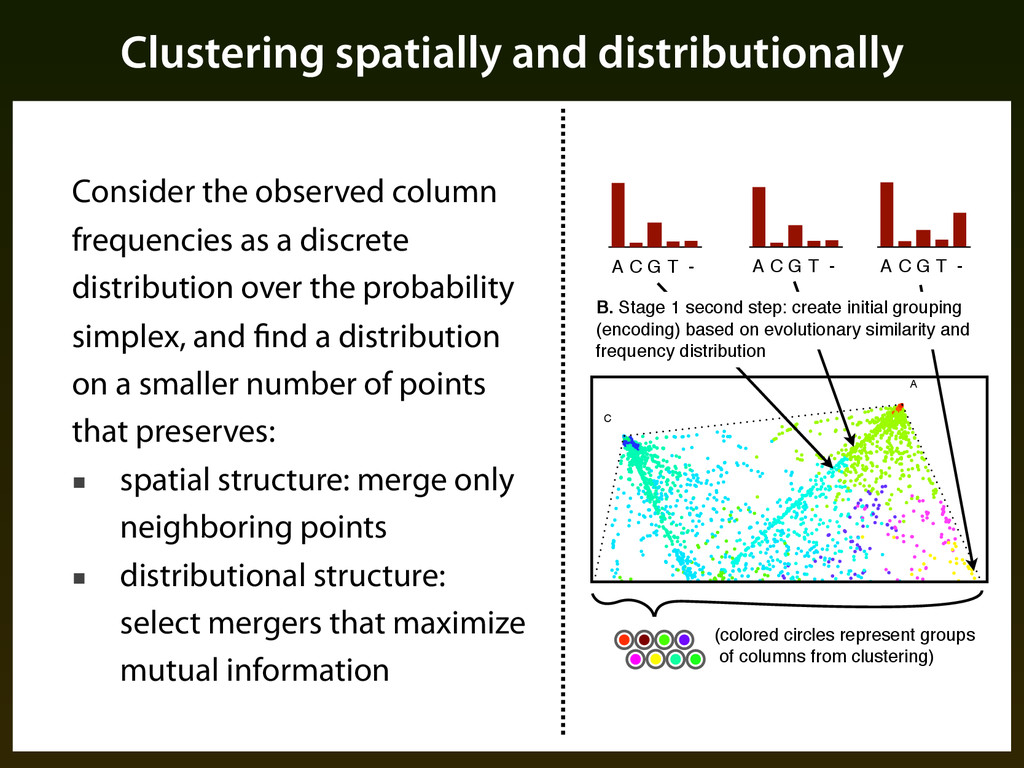
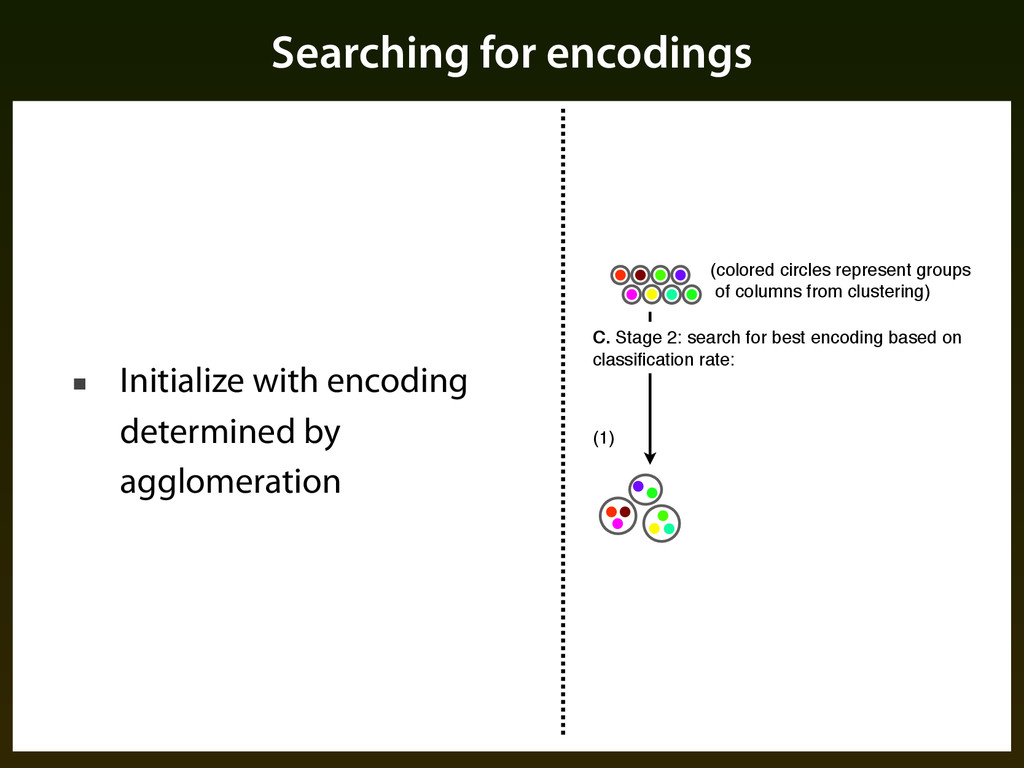
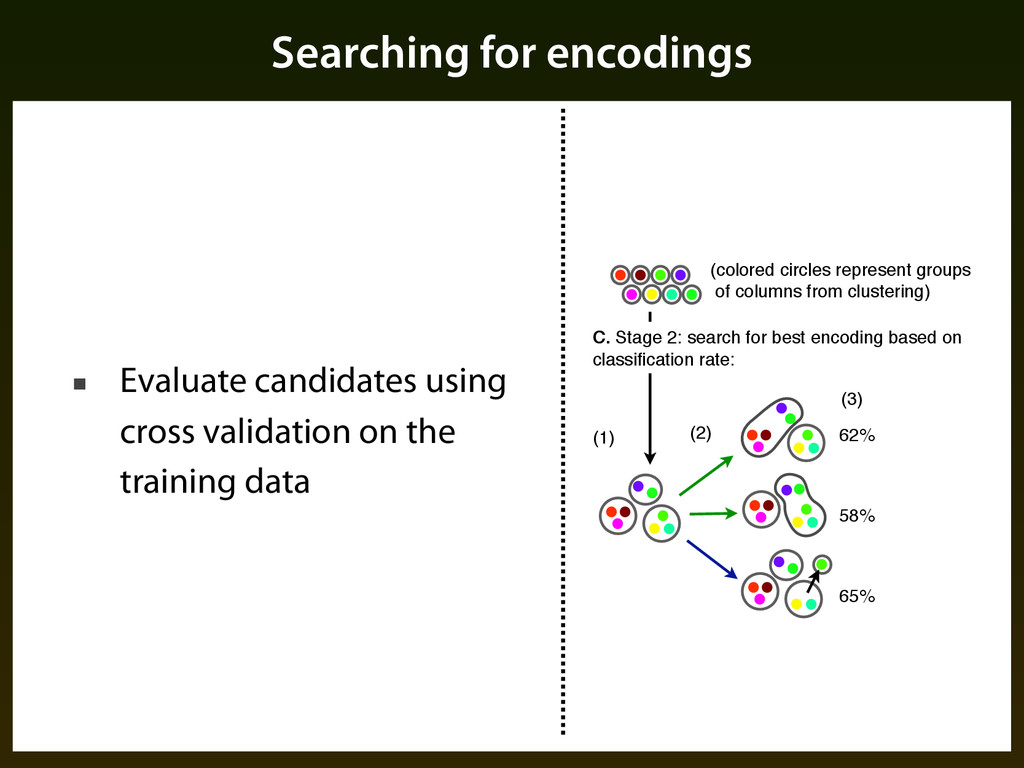
Create initial grouping of columns using an agglomeration procedures that combines evolutionary similarity with frequency distribution ▪ Search for final grouping of columns using iterative procedure guided by classification performance
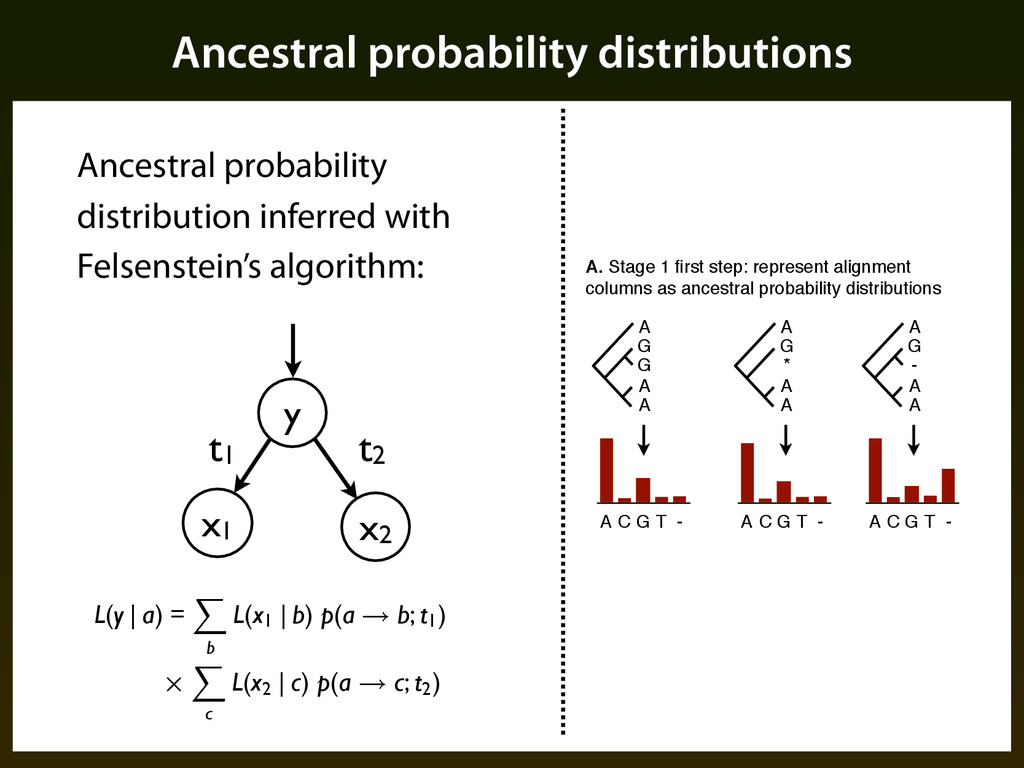
A G G A A A C G T - A G - A A A C G T - A G * A A A. Stage 1 first step: represent alignment columns as ancestral probability distributions A C G T - y x1 t1 t2 x2 L(y | a) = b L(x1 | b) p(a ⇥ b; t1 ) c L(x2 | c) p(a ⇥ c; t2 ) Q = ⇧ ⇧ ⇤ - a b c d - e f g h - i ⇥ ⌃ ⌃ ⌅ b) p(a ⇥ b; t1 ) c L(x2 | c) p(a ⇥ c; t2 ) - a b c ⇥
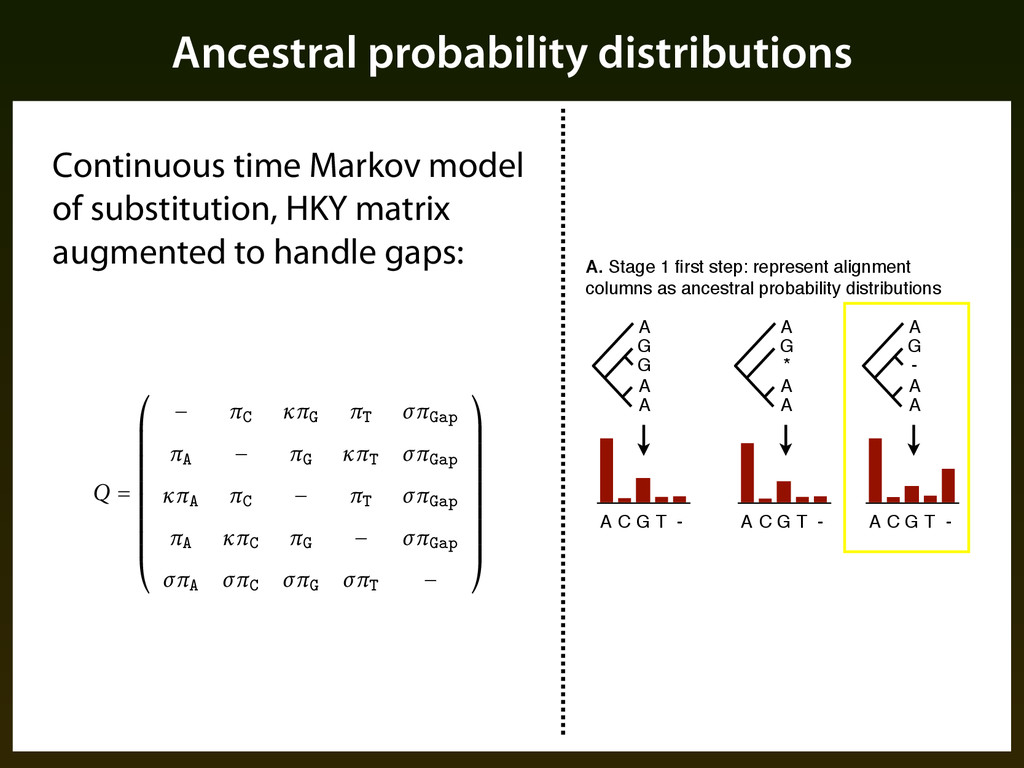
G T - A G - A A A C G T - A G * A A A. Stage 1 first step: represent alignment columns as ancestral probability distributions A C G T - Continuous time Markov model of substitution, HKY matrix augmented to handle gaps: nch of the phylogenetic tree. We assume a continuous time Markov process rate matrix Q speci⇠es the instantaneous rate of each substitution event, s the rates in Q through a smaller number of parameters. In particular, parameterization provided by the HKY model of Hasegawa et al. (⌧ ) of equilibrium probabilities for each base (⌫ parameters; ⌅ A , ⌅ C , ⌅ G , ⌅ T ), io between the rates of transitions and transversions (⇧)%. We extend to accommodate gaps as if they were a '(h nucleotide, introducing an equilibrium probability (⌅ Gap ) and rate ratio (gaps to transversions ⌃), e rate matrix: Q = ⇤ ⌥ ⌥ ⌥ ⌥ ⌥ ⌥ ⌥ ⌥ ⌥ ⌥ ⇧ ⌅ C ⇧⌅ G ⌅ T ⌃⌅ Gap ⌅ A ⌅ G ⇧⌅ T ⌃⌅ Gap ⇧⌅ A ⌅ C ⌅ T ⌃⌅ Gap ⌅ A ⇧⌅ C ⌅ G ⌃⌅ Gap ⌃⌅ A ⌃⌅ C ⌃⌅ G ⌃⌅ T ⌅ ⌃ ameters of Q are estimated using the Expectation Maximization algorithm ed in the PHAST so⌧ware package (Siepel and Haussler, ), generally d tree topology and a sample of genome-wide alignments.
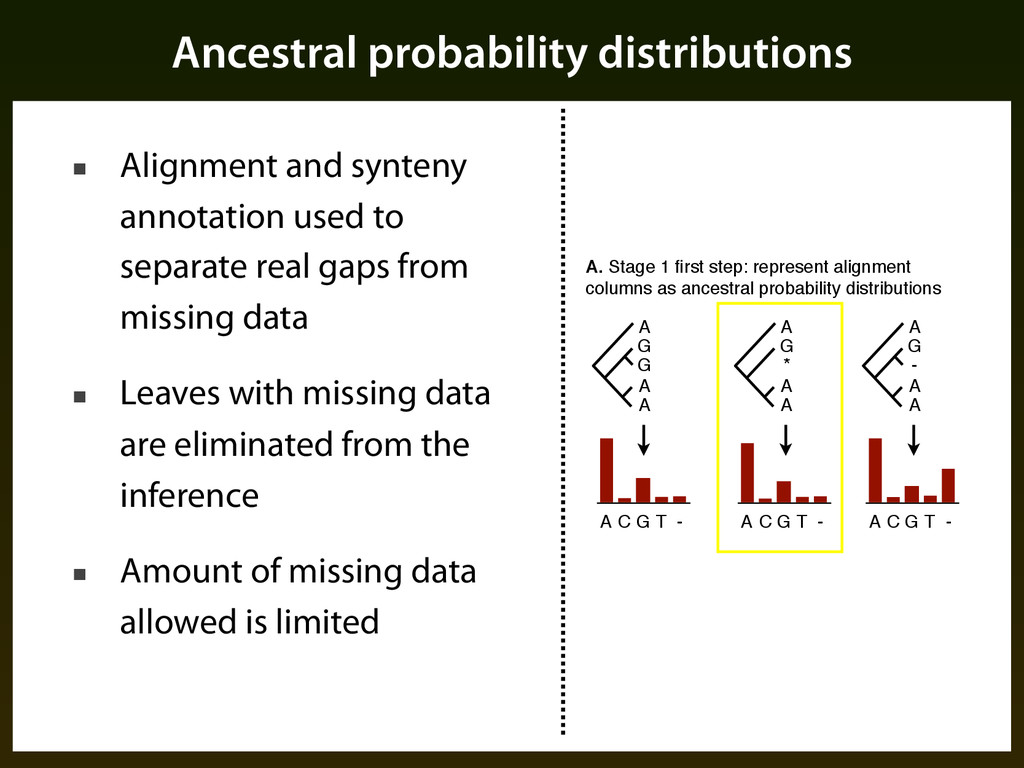
G T - A G - A A A C G T - A G * A A A. Stage 1 first step: represent alignment columns as ancestral probability distributions A C G T - ▪ Alignment and synteny annotation used to separate real gaps from missing data ▪ Leaves with missing data are eliminated from the inference ▪ Amount of missing data allowed is limited
▪ Centroid of a cluster is the average of all points in cluster weighted by their probabilities ▪ Distance / linkage is euclidean between centroids ▪ Iteration: ▪ Consider each point and its nearest neighbor ▪ Compute entropy if that pair were merged, keep merger that maximizes entropy
set of mergers and expansions from the current encoding (1) (2) (colored circles represent groups of columns from clustering) C. Stage 2: search for best encoding based on classification rate:
the training data 62% 58% 65% (1) (2) (3) (colored circles represent groups of columns from clustering) C. Stage 2: search for best encoding based on classification rate:
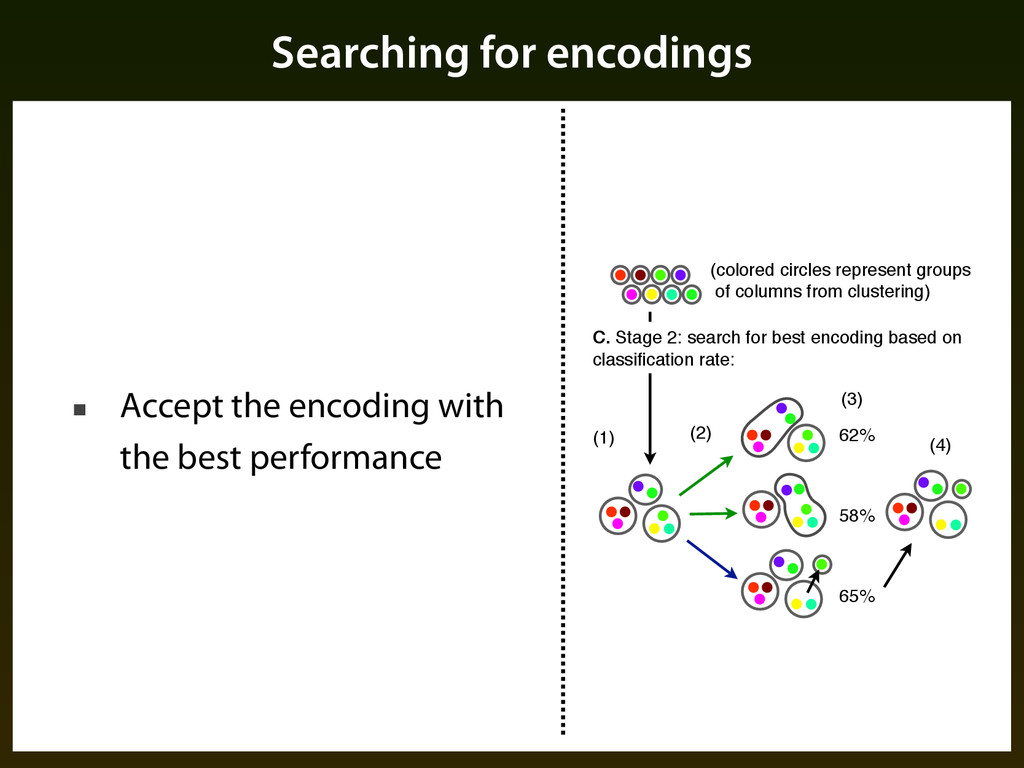
performance 62% 58% 65% (1) (2) (3) (4) (colored circles represent groups of columns from clustering) C. Stage 2: search for best encoding based on classification rate:
with no performance improvement) 62% 58% 65% (1) (2) (3) (4) (5) (colored circles represent groups of columns from clustering) C. Stage 2: search for best encoding based on classification rate:
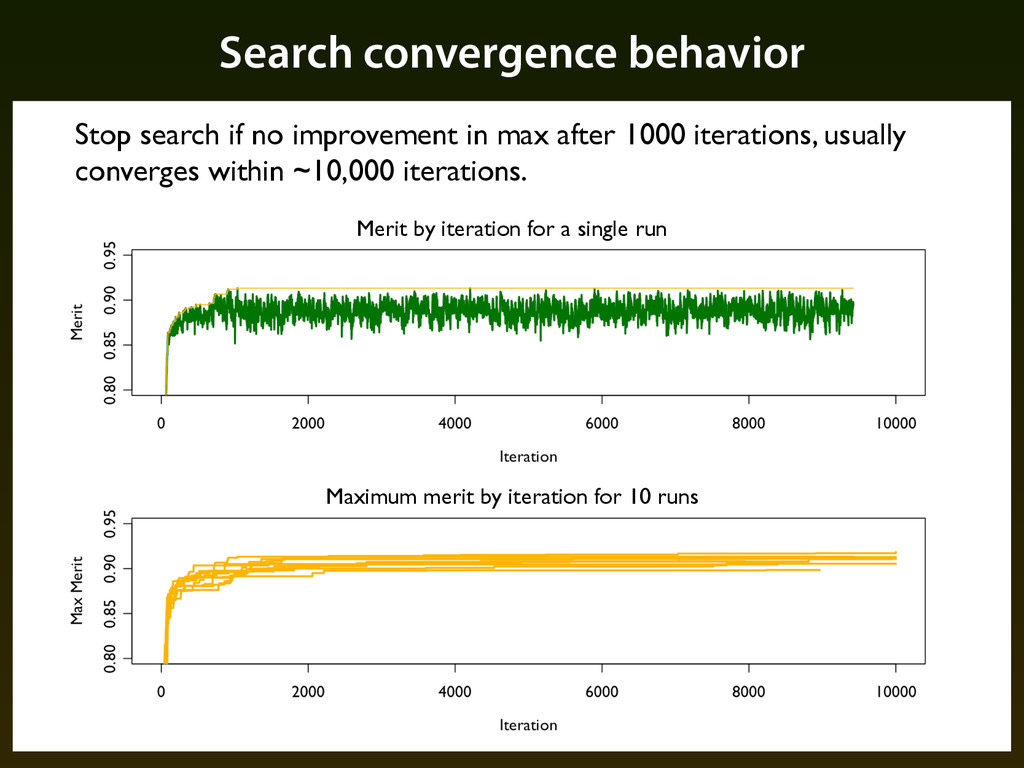
for smaller models the search can get stuck in local minima — we force several consecutive “expansion” steps if there is no improvement in performance for 20 iterations ▪ Given the large space of possible alphabets the search process can get lost — we restart from the best alphabet found so far if there is no improvement in performance for 50 iterations
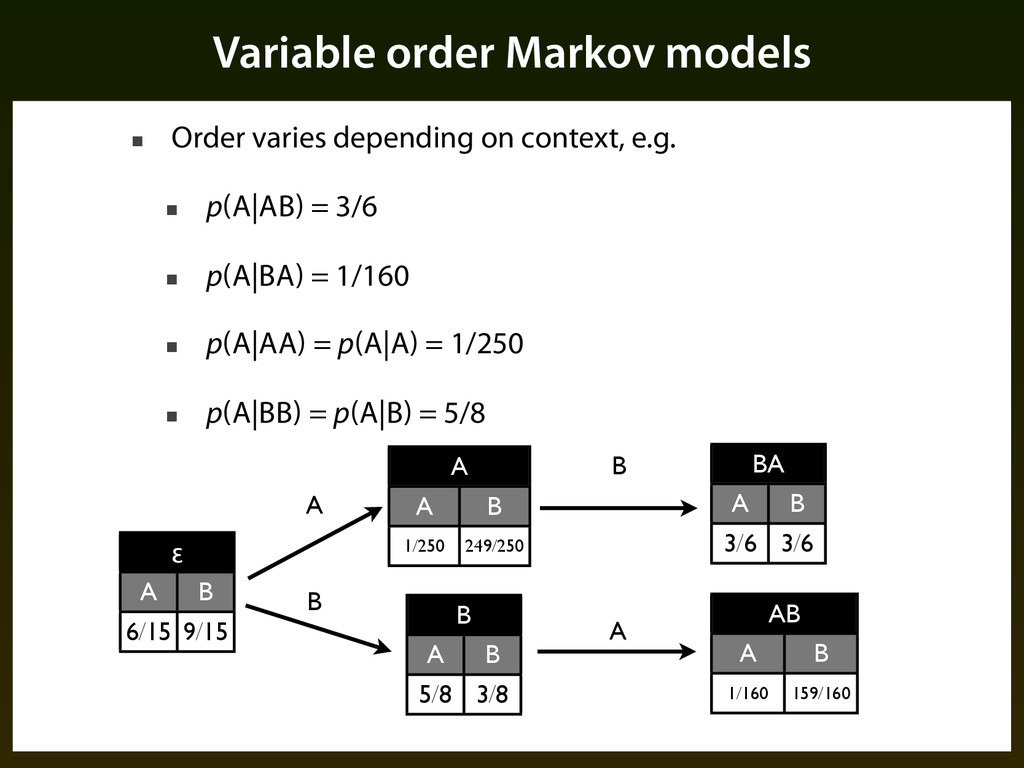
3/6 ▪ p(A|BA) = 1/160 ▪ p(A|AA) = p(A|A) = 1/250 ▪ p(A|BB) = p(A|B) = 5/8 Variable order Markov models BA BA A B 3/6 3/6 AB AB A B 1/160 159/160 ε ε A B 6/15 9/15 A A A B 1/250 249/250 B B A B 5/8 3/8 A A B B
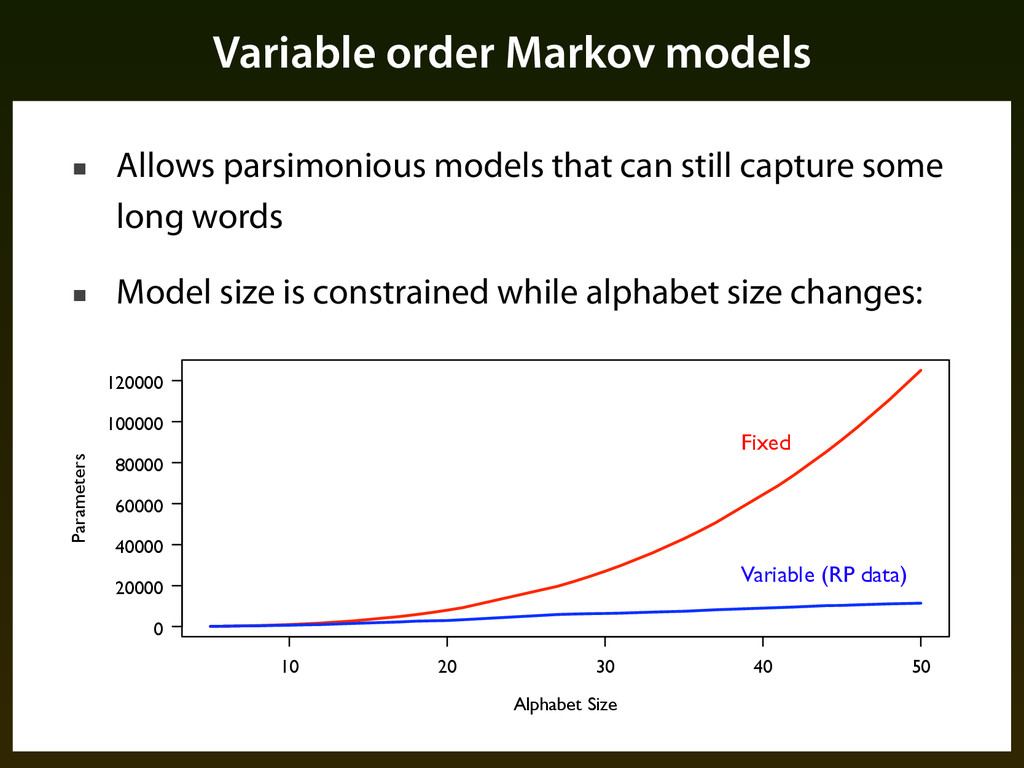
still capture some long words ▪ Model size is constrained while alphabet size changes: 10 20 30 40 50 0 20000 40000 60000 80000 100000 120000 Alphabet Size Parameters Fixed Variable (RP data)
“known regulatory” from “neutral” regions ▪ Seven species alignments: human, chimpanzee, macaque, mouse, rat, dog, cow ▪ Training data sets of ~31,000 bases each (no more than three missing species allowed in a column) ▪ 17 symbol final alphabet ▪ Cross validation success rate (leave-one-out) of ~94%, a substantial improvement over ~82% for three-species RP
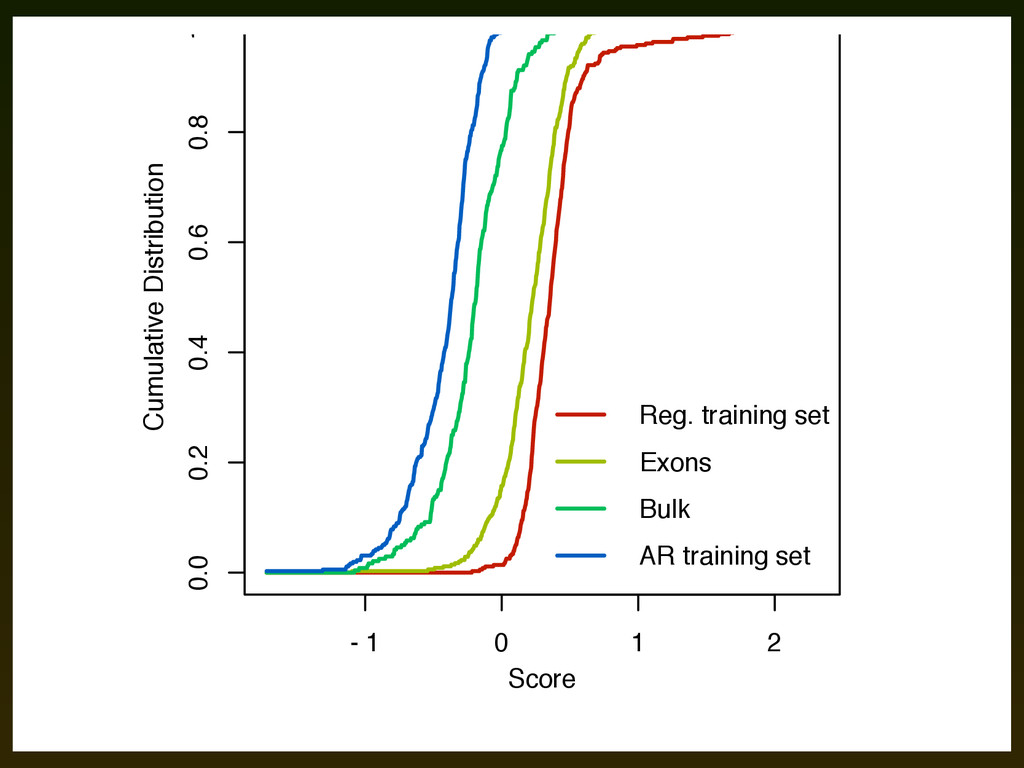
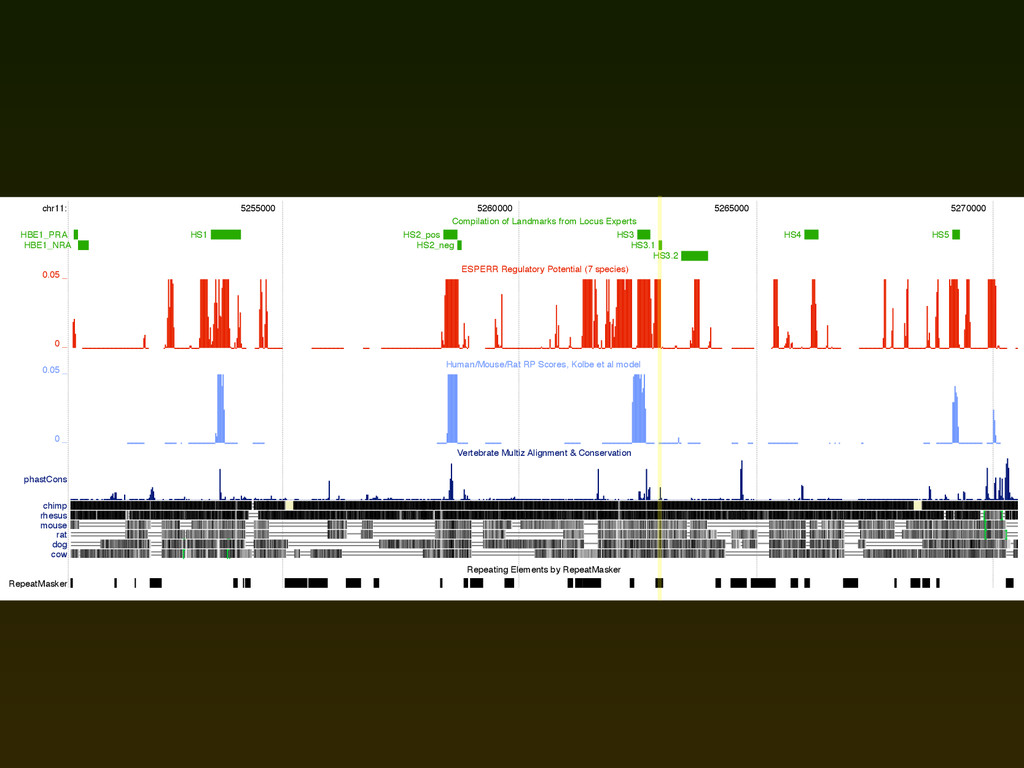
experimentally confirmed regulatory elements in this region ▪ ROC plot considers the sensitivity/specificity of several scores for discriminating these elements
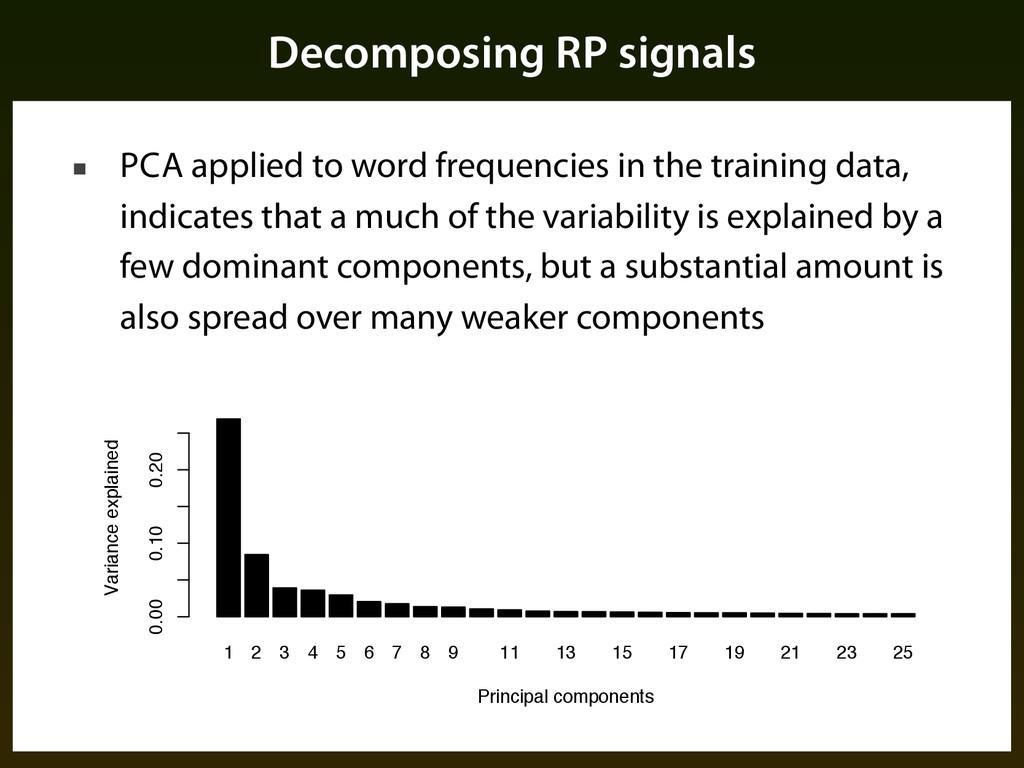
the training data, indicates that a much of the variability is explained by a few dominant components, but a substantial amount is also spread over many weaker components 1 2 3 4 5 6 7 8 9 11 13 15 17 19 21 23 25 Principal components Variance explained 0.00 0.10 0.20 p. .6 RP
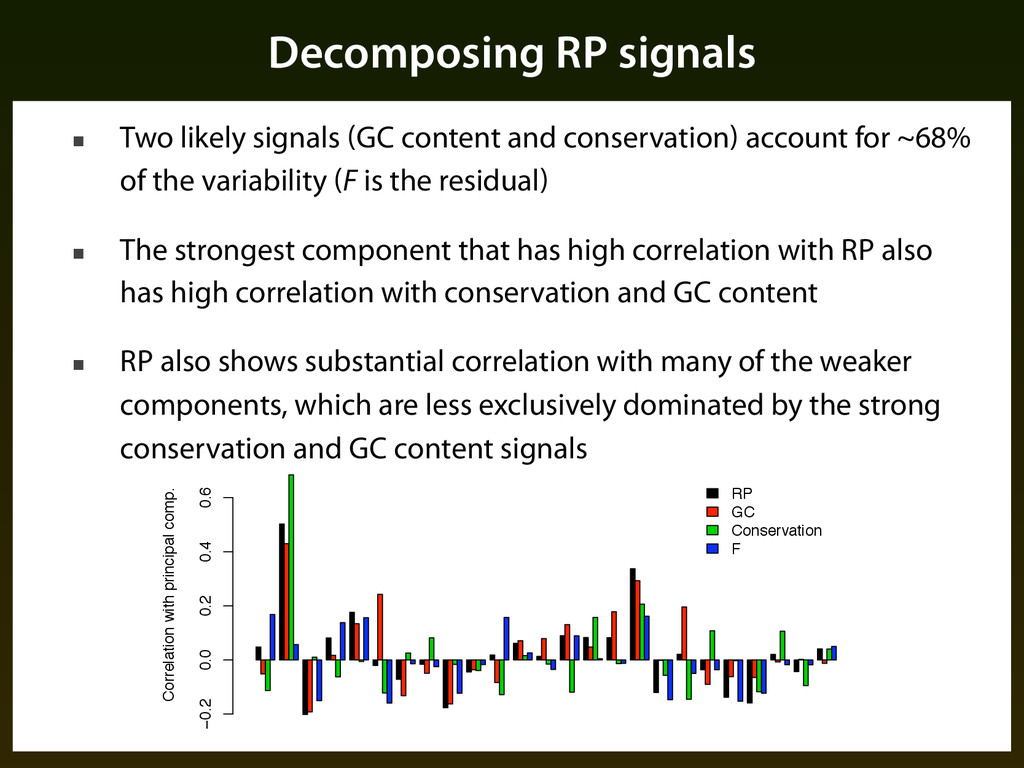
conservation) account for ~68% of the variability (F is the residual) ▪ The strongest component that has high correlation with RP also has high correlation with conservation and GC content ▪ RP also shows substantial correlation with many of the weaker components, which are less exclusively dominated by the strong conservation and GC content signals 1 2 3 4 5 6 7 8 9 11 13 15 17 19 21 23 25 Principal components Variance explained 0.00 0.10 0.20 Correlation with principal comp. −0.2 0.0 0.2 0.4 0.6 RP GC Conservation F
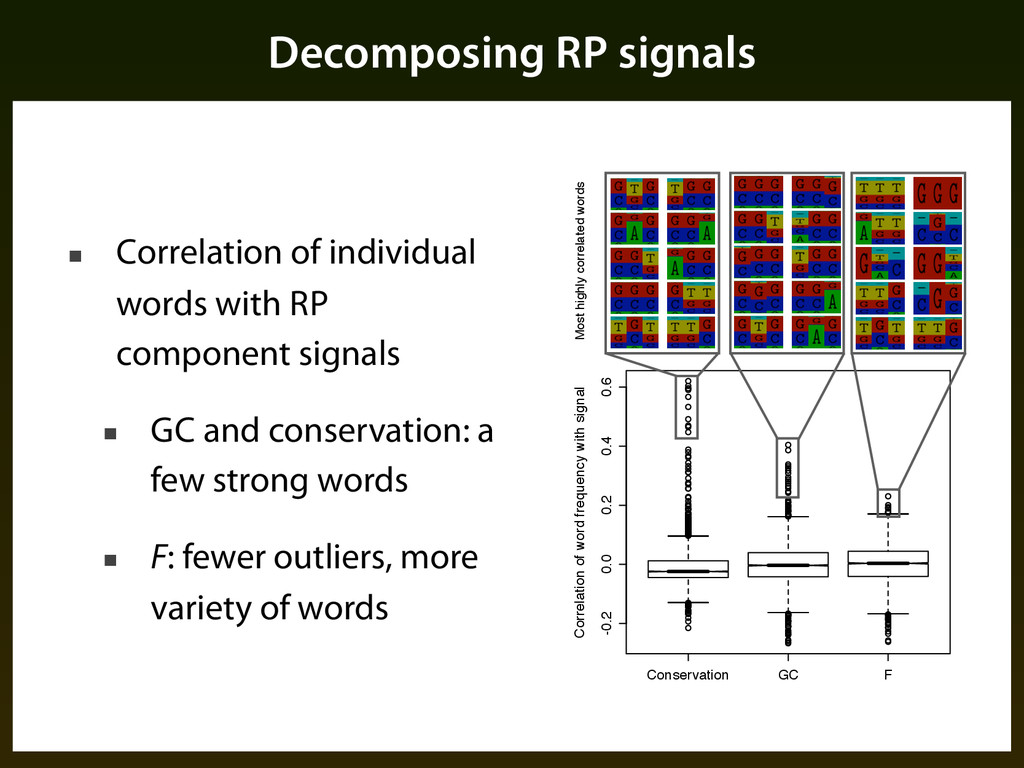
component signals ▪ GC and conservation: a few strong words ▪ F: fewer outliers, more variety of words Figure %.%: Share of variance explained by each of the #rst $& principal the RP training data word frequencies (top) and correlation of RP sco conservation, and the residuals F with each principal component (b Conservation GC F -0.2 0.0 0.2 0.4 0.6 Correlation of word frequency with signal Most highly correlated words 05/29/2 http://www.bx.psu.edu/~james/rp_2006/logos_7way/high_cons.html http://www.bx.psu.edu/~james/rp_2006/logos_7way/high_cons.html http://www.bx.psu.edu/~james/rp_2006/logos_7way/high_gc.html http://www.bx.psu.edu/~james/rp_2006/logos_7way/high_gc.html http://www.bx.psu.edu/~james/rp_2006/logos_7way/high_F.ht http://www.bx.psu.edu/~james/rp_2006/logos_7way/high_F.ht Figure ⇡.⇢: Distributions of the correlations between word frequen
ENCODE data ▪ Defined by a sequence specific ChIP-chip hit ▪ Supported by secondary evidence ▪ More than 2.5kb away from a TSS ▪ Elements with high GC / conservation associated with predicted promoters and other promoter-like characteristics ▪ Elements with high F show much less associated with these characteristics: more likely to be distal
marker for regulatory function ▪ ENCODE comprehensively tested 1% of the genome for this feature ▪ We derive from their data a stringent set of positive and negative examples
2005) found that a linear SVM trained on word frequencies could discriminate such sites, however on this dataset applying their approach achieves only 60% success (leave-one-out cross validation) ▪ ESPERR identifies an 18 symbol encoding which achieves a success rate of ~80%
(~143kb) and 138 non-validated (~165kb); all tested regions are highly conserved ▪ Alignments of human, mouse, opossum, chicken, frog, zebrafish and pufferfish ▪ ESPERR identifies an encoding to 15 symbols that achieves ~83% cross validation success rate
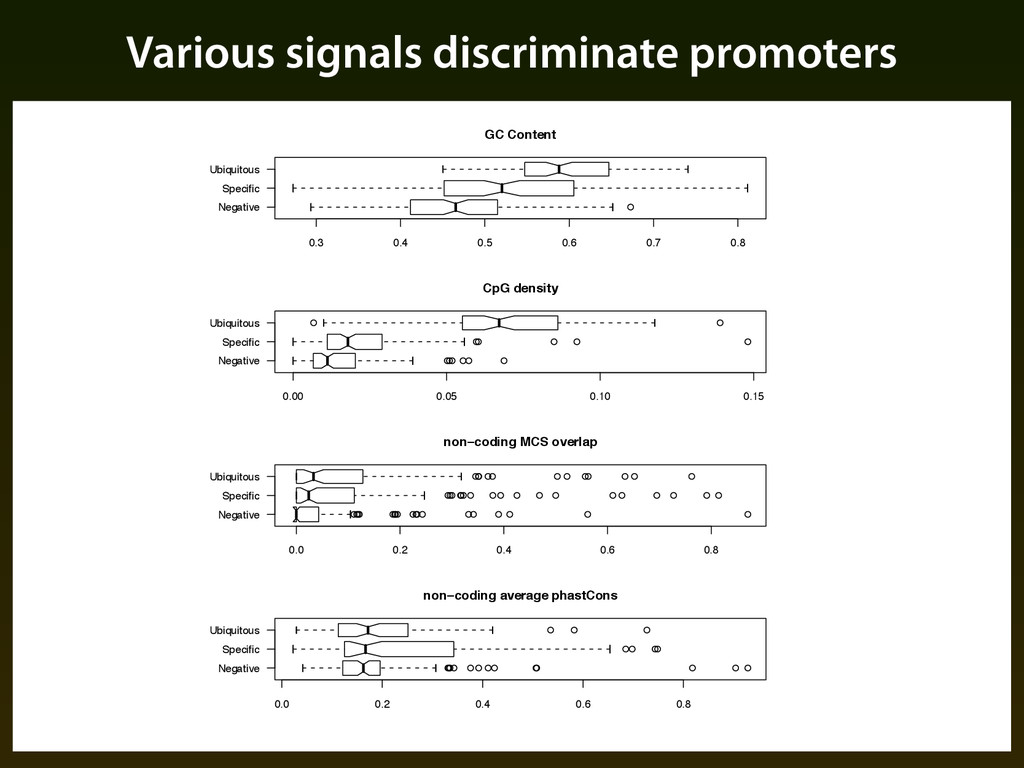
et al. (2006) tested promoter activity at the 5’ ends of aligned cDNAs in the ENCODE regions ▪ Most regions tested in 16 cell lines, from these we derive three training sets: ▪ “Ubiquitous” — positive in all 16 cell lines (106) ▪ “Specific” — positive in 1 to 5 cell lines (130) ▪ “Negative” — negative in all 16 cell lines (123)
to discriminate each pair of training sets using: ▪ ESPERR with five species alignments (human, chimpanzee, mouse, rat, dog) ▪ Naive-bayes classification using four other quantities: GC content, CpG content, MCS overlap, and phastCons. ▪ Leave-one-out cross validation used for all cases
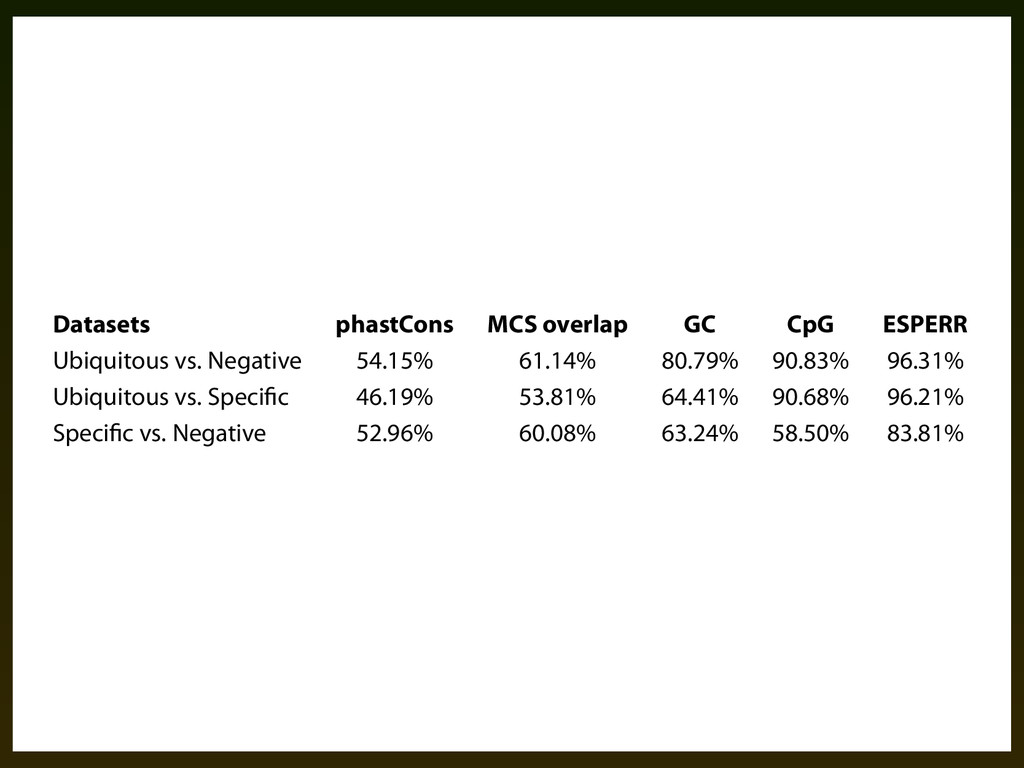
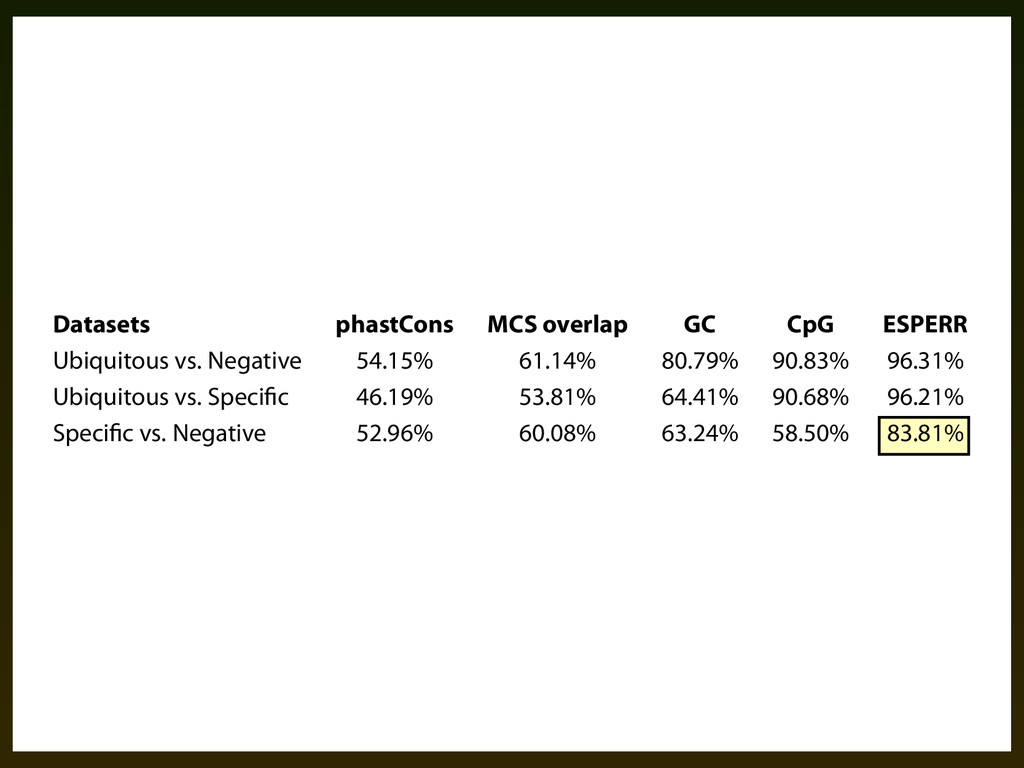
(canFam-) to the human genome. Positions overlapping coding sequences were eliminated from the training data. We allowed any column in the training data with at most two missing species to be used. Handling missing data in this way allowed us to cover most potential promoter regions, however a small number of training regions (+ Datasets phastCons MCS overlap GC CpG ESPERR Ubiquitous vs. Negative 54.15% 61.14% 80.79% 90.83% 96.31% Ubiquitous vs. Speci c 46.19% 53.81% 64.41% 90.68% 96.21% Speci c vs. Negative 52.96% 60.08% 63.24% 58.50% 83.81% Table .⌥. Pair-wise classi)cation success rates using quantities computed from genomic sequence (GC content and CpG density), alignments (phastCons and MCS overlap), and ESPERR.
(canFam-) to the human genome. Positions overlapping coding sequences were eliminated from the training data. We allowed any column in the training data with at most two missing species to be used. Handling missing data in this way allowed us to cover most potential promoter regions, however a small number of training regions (+ Datasets phastCons MCS overlap GC CpG ESPERR Ubiquitous vs. Negative 54.15% 61.14% 80.79% 90.83% 96.31% Ubiquitous vs. Speci c 46.19% 53.81% 64.41% 90.68% 96.21% Speci c vs. Negative 52.96% 60.08% 63.24% 58.50% 83.81% Table .⌥. Pair-wise classi)cation success rates using quantities computed from genomic sequence (GC content and CpG density), alignments (phastCons and MCS overlap), and ESPERR.
agnostic, all that is needed is to change the underlying classifier ▪ Rather than a log-odds score, we train a VOMM for each class and assign elements to the model under which they have the highest probability ▪ Figure of merit remains the fraction of elements correctly classified under cross validation
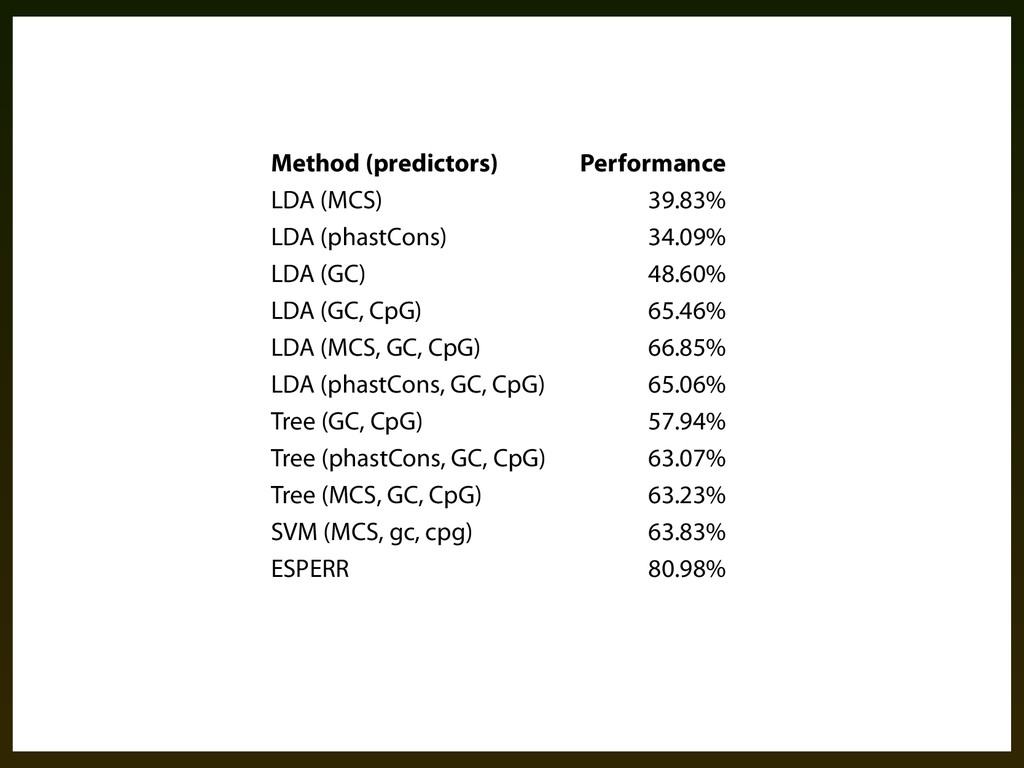
other multi-way classifiers using various combinations of signals: ▪ LDA (linear discriminant analysis) ▪ Classification trees (RPART) ▪ SVM (various kernels)
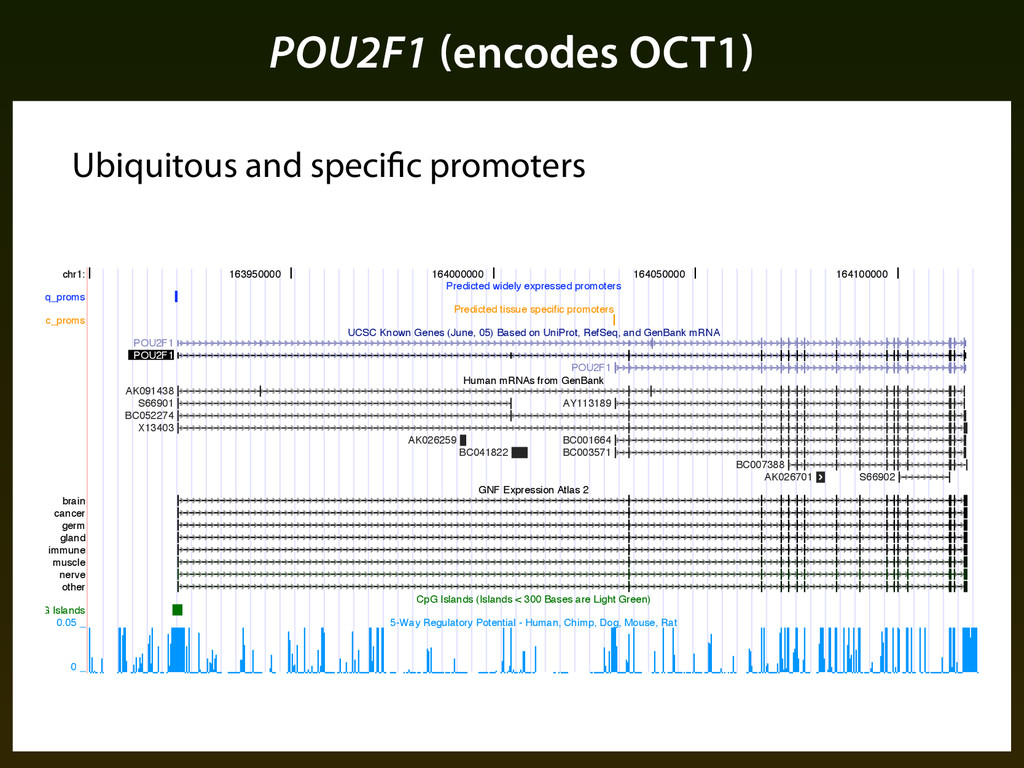
Cooper et al. (based on 5’ ends of cDNA alignments) we identified 79,616 potential promoter regions genome wide ▪ Using the encoding determined by ESPERR we predicted whether each potential promoter would be ubiquitously expressed (~19k), specifically expressed (~23k), or not active (~29k). ▪ We find ~6,000 gene models (clusters of overlapping cDNA alignments) with both a specific and ubiquitous promoter
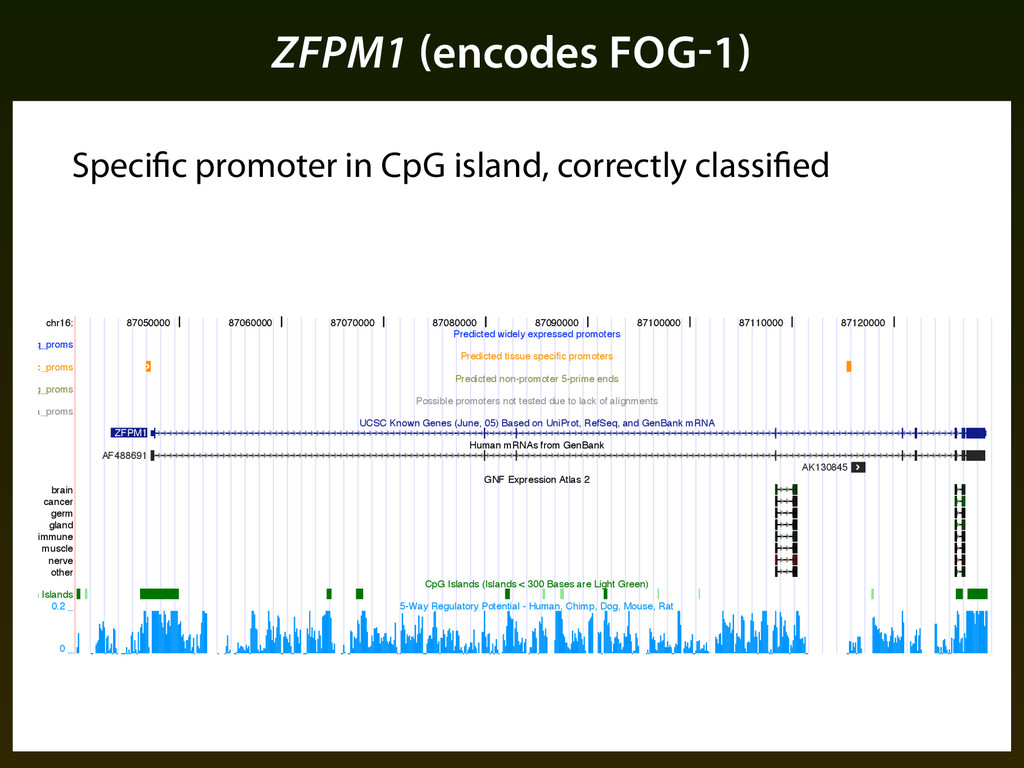
germ gland immune muscle nerve other CpG Islands 87050000 87060000 87070000 87080000 87090000 87100000 87110000 87120000 Predicted widely expressed promoters Predicted tissue specific promoters Predicted non-promoter 5-prime ends Possible promoters not tested due to lack of alignments UCSC Known Genes (June, 05) Based on UniProt, RefSeq, and GenBank mRNA Human mRNAs from GenBank GNF Expression Atlas 2 CpG Islands (Islands < 300 Bases are Light Green) 5-Way Regulatory Potential - Human, Chimp, Dog, Mouse, Rat ZFPM1 AF488691 AK130845 0.2 _ 0 _ Figure ⌧.⇢: UCSC genome browser snapshot of promoter predictions in the neighborhood of ZFPM⇧. Specific promoter in CpG island, correctly classified
a variety of problems ▪ Can capture combinations of many signals ▪ Can be used with different underlying classifiers for pairwise and multi-way classification
high throughput experimental assays for protein binding (like ChIP-chip) ▪ Interpreting encodings and understanding the specific signals captured for a given problem ▪ Better modeling of indels ▪ Moving RP beyond mammals ▪ Other classifiers... gene prediction
signals in genomic sequence alignments to identify functional elements”, written with Svitlana Tyekucheva, David King, Ross Hardison, Webb Miller and Francesca Chiaromonte ▪ “Leveraging ENCODE data to predict widely expressed and tissue-specific transcriptional promoters in the human genome”, written with Nathan Trinklein, Ross Hardison, Webb Miller and Francesca Chiaromonte ▪ ENCODE consortium, the CCGB
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}