C.S.; Stephan, K.E.; Buhmann, J.M. (2010). The balanced accuracy and its posterior distribution. Proceedings of the 20th International Conference on Pattern Recognition, 3121-24. 多クラス分類問題にも適用可能。ランダムな分類モデルと比較 した際にどれだけパフォーマンスが良いかを示す指標 Pr(a): 観察された一致率 Pr(e): 期待一致率 McHugh ML. Interrater reliability: the kappa statistic. Biochem Med (Zagreb). 2012;22(3):276-82. Landis, J.R.; Koch, G.G. (1977). “The measurement of observer agreement for categorical data”. Biometrics 33 (1): 159–174
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
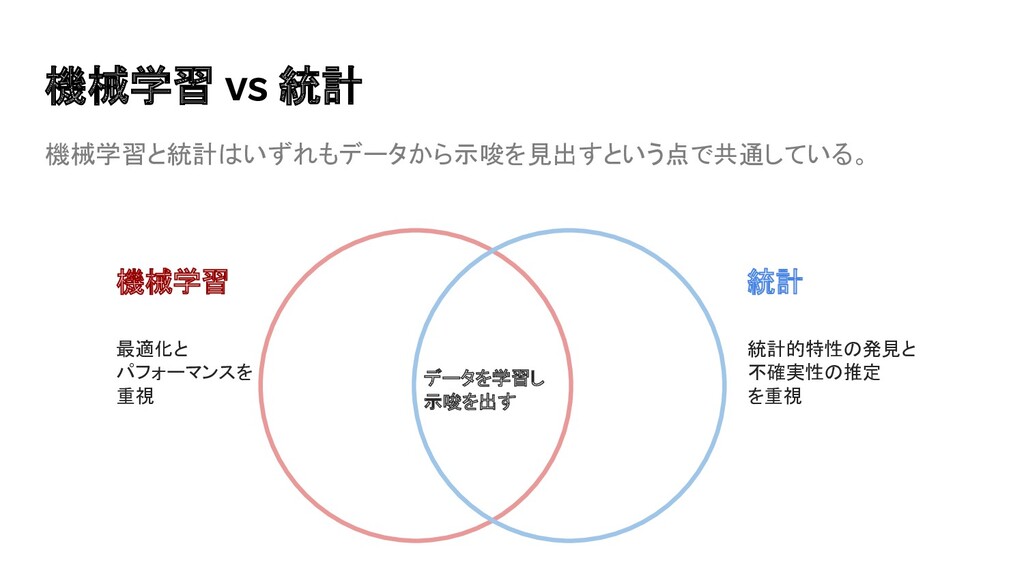{kind=link}
{kind=link}
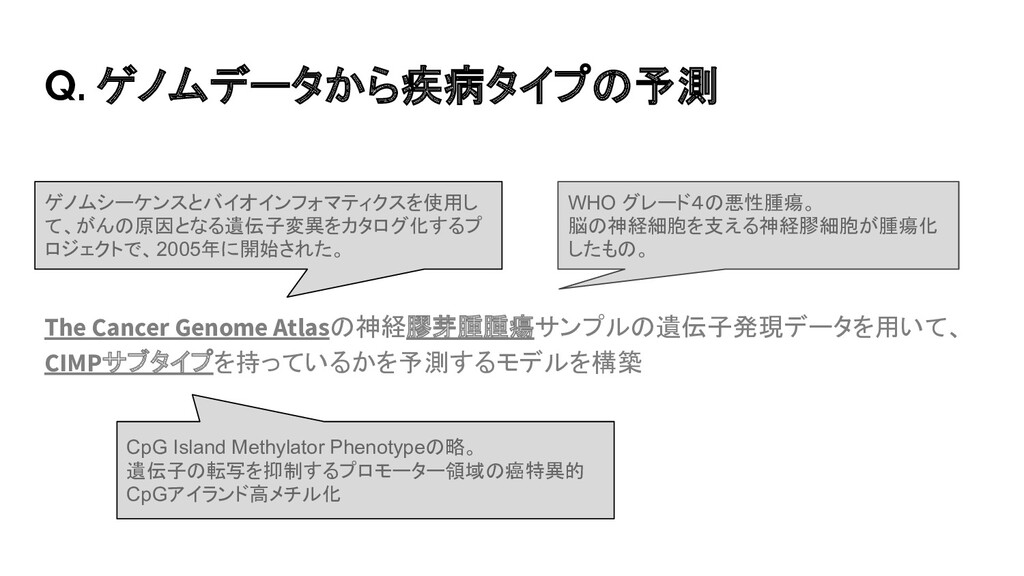{kind=link}
{kind=link}
![実験における測定誤差やデータ取得の仮定で生じる潜在的な問題により、データスケー ルに違いが生じていないか箱ひげ図により確認する。 理想的には、各腫瘍サンプルは遺伝子発現値の同様の分布を持っている。 データ前処理(正規化) boxplot(gexp[,1:50],outline=FALSE,col="cornflowerblue" ) サンプルあたりの遺伝子発現値は同じス ケールとなっており、正規化が既に行われ たと考えられる 最初の50個の腫瘍](https://files.speakerdeck.com/presentations/3618b7d86e7140b28d45e1ede02c8de2/slide_9.jpg){kind=link}
![対数変換により扱いやすい分布に変換する。 データ前処理(変換) par(mfrow=c(1,2)) # 5番目の患者について、遺伝子発現量の分布を可視化する hist(gexp[,5],xlab="gene expression" ,main="", border="blue4", col="cornflowerblue"](https://files.speakerdeck.com/presentations/3618b7d86e7140b28d45e1ede02c8de2/slide_10.jpg){kind=link}
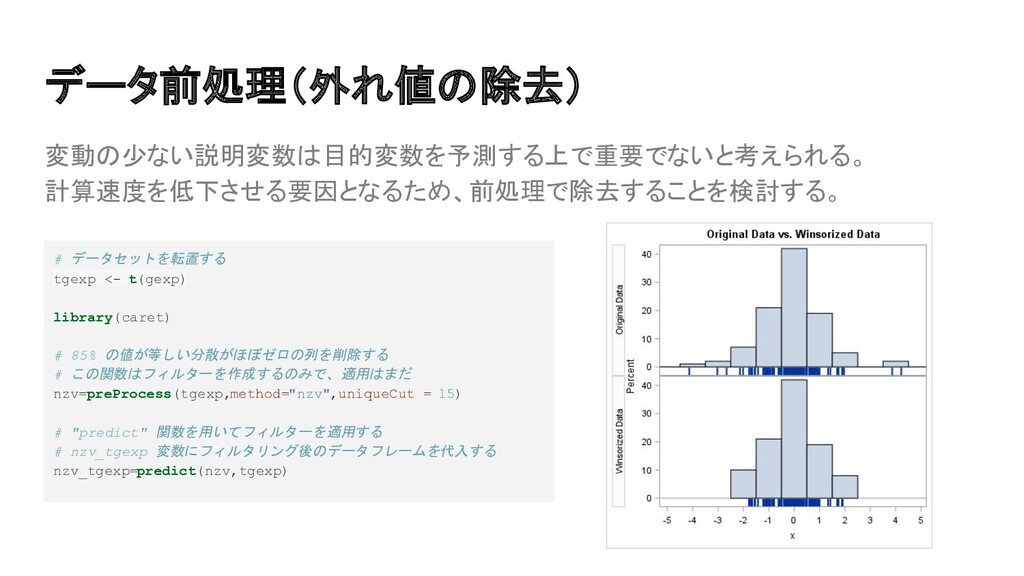{kind=link}
![変動の大きさについて、任意のカットオフ値を選択することもできる。 下記では、上位1000個の変数予測子を選択する。 データ前処理(外れ値の除去) par(mfrow=c(1,2)) # 5番目の患者について、フィルタリング前の遺伝子発現量の分布を可視化する hist(tgexp[,5],xlab="gene expression" ,main="", border="blue4",](https://files.speakerdeck.com/presentations/3618b7d86e7140b28d45e1ede02c8de2/slide_12.jpg){kind=link}
![変動の大きさについて、任意のカットオフ値を選択することもできる。 標準化により、全ての予測変数の平均は0、標準偏差は1になる。 この処理は通常、一部の計算の数値安定性を向上させるために使用される。 データ前処理(標準化) par(mfrow=c(1,2)) # 5番目の患者について、標準化前の遺伝子発現量の分布を可視化する hist(tgexp[,5],xlab="gene expression" ,main="",](https://files.speakerdeck.com/presentations/3618b7d86e7140b28d45e1ede02c8de2/slide_13.jpg){kind=link}
{kind=link}
![実験中の技術的な問題により、特定の遺伝子やゲノム位置の値がない場合(欠落値) が起こり得る。中央値や平均値、最近傍で補完することが一般的。 データ前処理(欠損値除去) # 欠損値が存在するデータを生成 missing_tgexp=tgexp missing_tgexp[ 1,1]=NA anyNA(missing_tgexp) #](https://files.speakerdeck.com/presentations/3618b7d86e7140b28d45e1ede02c8de2/slide_15.jpg){kind=link}
{kind=link}
{kind=link}
![最も簡単な推定手法として、類似したサンプルのラベルと同一のラベルを割り当てること が考えられる。概念的にはクラスタリングに近い。 Rではcaret::knn3()メソッドでk最近傍法を実行することができる。 k最近傍法によるCIMPサブタイプの推定 library(caret) knnFit=knn3(x=training[,-1], # 学習データ y=training[,1], #](https://files.speakerdeck.com/presentations/3618b7d86e7140b28d45e1ede02c8de2/slide_18.jpg){kind=link}
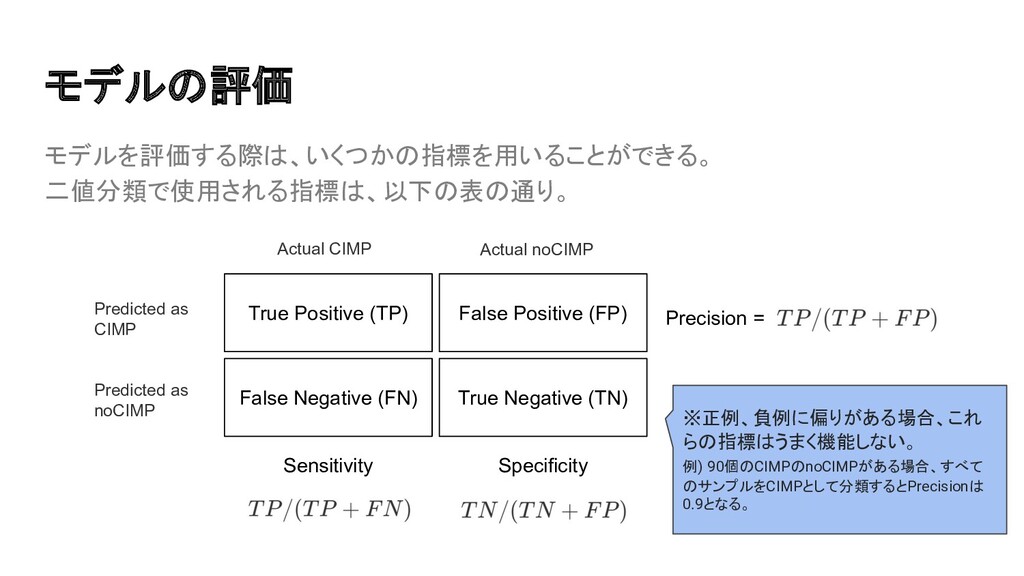{kind=link}
{kind=link}
![Rでモデルの各指標を確認するには、 caret :: confusionMatrix()関数を用いる モデルの評価 # 学習データに対して、 k=5のk-NN 実行結果を取得 knnFit=knn3(x=training[,-1],](https://files.speakerdeck.com/presentations/3618b7d86e7140b28d45e1ede02c8de2/slide_21.jpg){kind=link}
![テストデータについても同様に 精度指標を取得する。 モデルの評価 # 学習データに対して推測を実行 testPred=predict(knnFit,testing[, -1],type="class") # 実際の正解ラベルと推測されたラベルを比較して精度指標を取得 confusionMatrix(data=testing[,1],reference=testPred)](https://files.speakerdeck.com/presentations/3618b7d86e7140b28d45e1ede02c8de2/slide_22.jpg){kind=link}
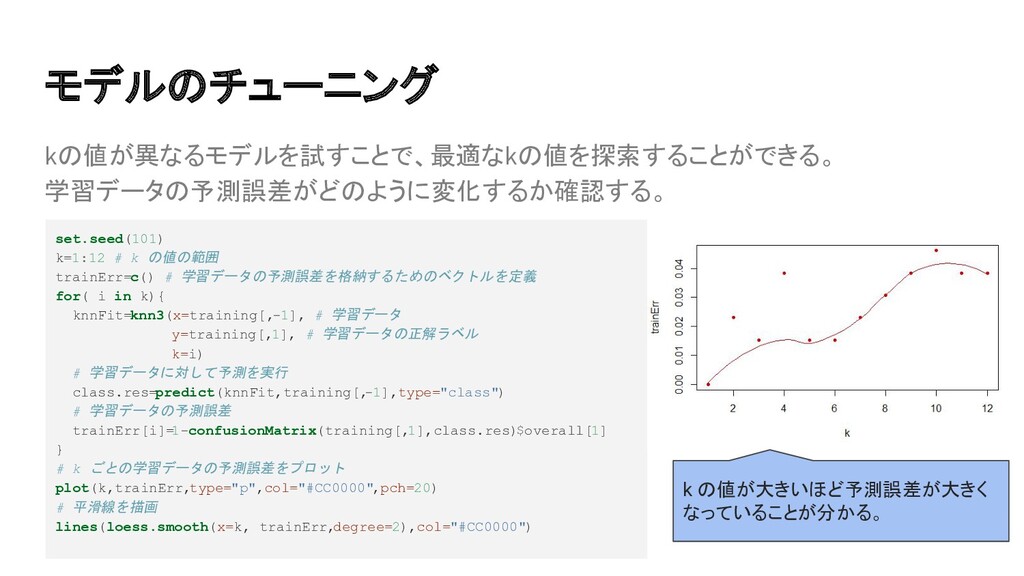{kind=link}
![テストデータについても、kの値で予測誤差がどのように変化するか確認する。 モデルのチューニング trainErr=c() # テストデータの予測誤差を格納するためのベクトルを定義 for( i in k){ knnFit=knn3(x=training[,-1],](https://files.speakerdeck.com/presentations/3618b7d86e7140b28d45e1ede02c8de2/slide_24.jpg){kind=link}
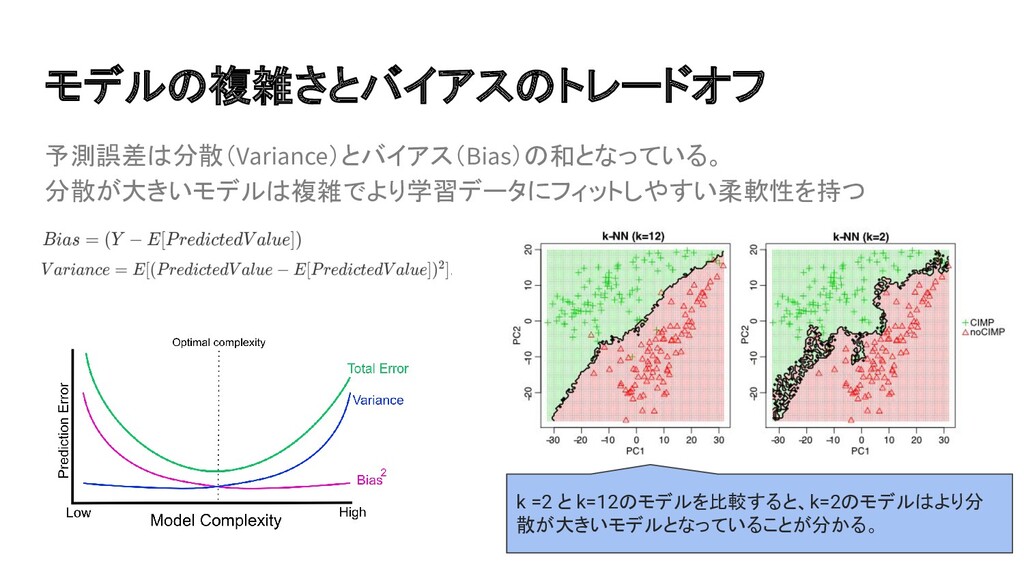{kind=link}
{kind=link}
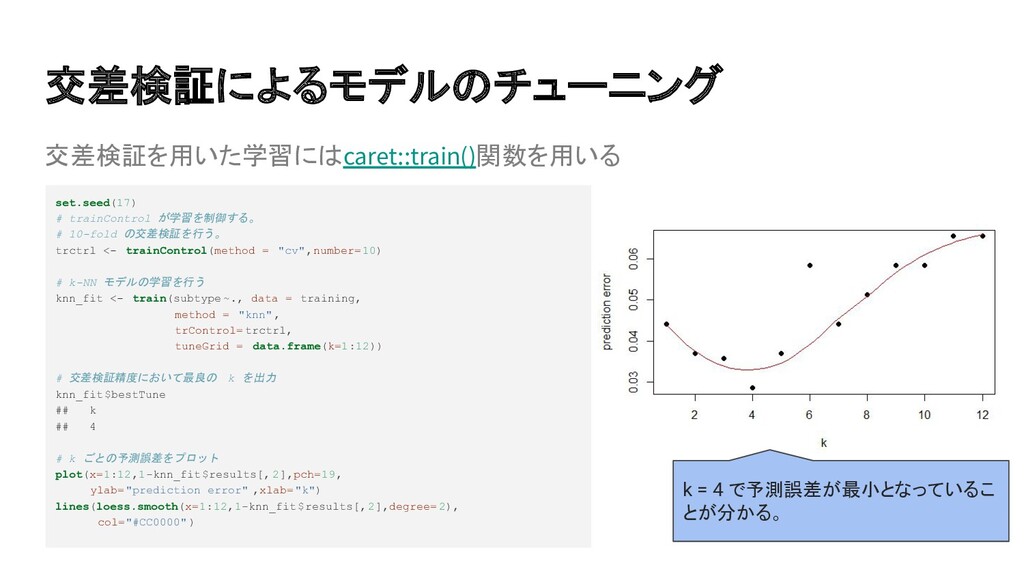{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}