# 平均値の差を計算 org.diff=mean(gene1)- mean(gene2) gene.df=data.frame(exp=c(gene1,gene2), group=c(rep("test",30),rep("control",30))) # 帰無分布を取得 exp.null <- do(1000) * diff(mosaic::mean(exp ~ shuffle(group), data=gene.df)) # ヒストグラムを描画 hist(exp.null[, 1],xlab="null distribution | no difference in samples" , main=expression(paste(H[0]," :no difference in means" )), xlim=c(-2,2),col="cornflowerblue" ,border="white") abline(v=quantile(exp.null[, 1],0.95),col="red" ) abline(v=org.diff,col="blue" ) text(x=quantile(exp.null[, 1],0.95), y=200, "0.05",adj=c(1,0), col="red") text(x=org.diff,y=200,"org. diff." ,adj=c(1,0),col="blue") Rでの実装 27
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
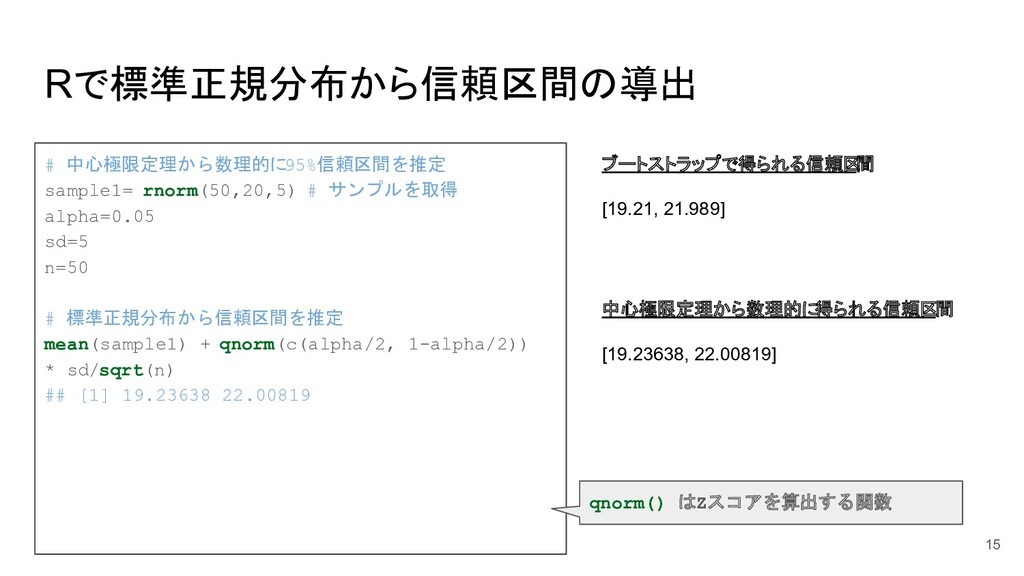
![Rでt分布から信頼区間の導出 ブートストラップで得られる信頼区 間 [19.21, 21.989] # 中心極限定理から数理的に 95%信頼区間を推定 sample1= rnorm(50,20,5)](https://files.speakerdeck.com/presentations/5c28601d336842c385d87a2fc3086388/slide_16.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Rでの実装 # P値(実際の平均値の差より高くなる確率)の算出 p.val=sum(exp.null[,1]>org.diff)/length(exp.null[,1]) p.val ## [1] 0.001 28](https://files.speakerdeck.com/presentations/5c28601d336842c385d87a2fc3086388/slide_27.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
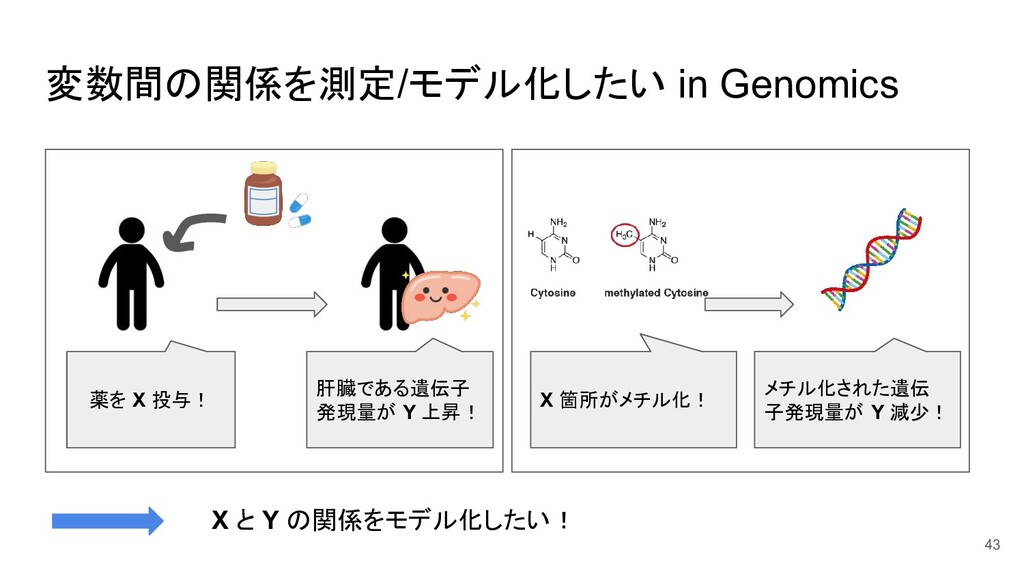{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
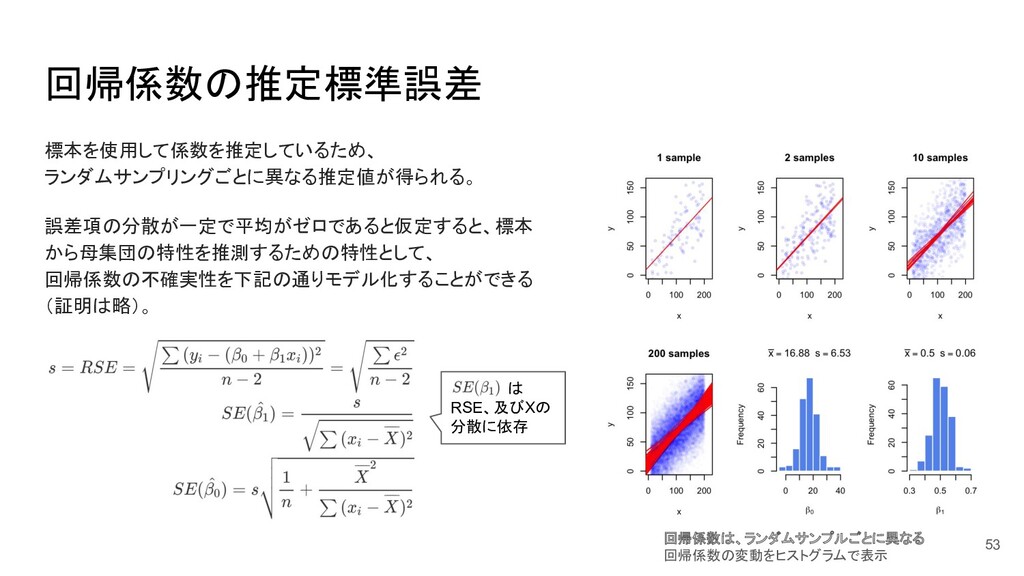{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}