Upgrade to Pro
— share decks privately, control downloads, hide ads and more …
Speaker Deck
Features
Speaker Deck
PRO
Sign in
Sign up for free
Search
Search
【第5回】ゼロから始めるゲノム解析(Python編)
Search
nkimoto
December 23, 2021
Technology
760
0
Share
Embed
Copy iframe code
Copy JS code
Copy link
Start on current slide
【第5回】ゼロから始めるゲノム解析(Python編)
2021/12/23 (木) 【第5回】ゼロから始めるゲノム解析(Python編) 資料
nkimoto
December 23, 2021
More Decks by nkimoto
See All by nkimoto
Location Restriction Sites: Using, Testing, and Sharing Code
nkimoto
0
380
Finding a Protein Motif: Fetching Data and Using Regular Expressions
nkimoto
0
350
Overlap Graphs: Sequence Assembly Using Shared K-mers
nkimoto
0
240
Computing GC Content: Parsing FASTA and Analyzing Sequences
nkimoto
0
330
【第3回】ゼロから始めるゲノム解析(Python編)
nkimoto
0
470
【第1回】ゼロから始めるゲノム解析(Python編).pdf
nkimoto
0
940
【第7回】ゼロから始めるゲノム解析.pdf
nkimoto
0
490
【第5回】ゼロから始めるゲノム解析(R編)
nkimoto
0
600
【第3回】ゼロから始めるゲノム解析(R編)
nkimoto
0
1.6k
Other Decks in Technology
See All in Technology
生成 AI 時代にいま一度「問い合わせ」について考えてみる
kazzpapa3
1
130
プロダクト開発組織の現在地(Ver.2026/07) / product-organization
kaonavi
0
400
AI時代の開発生産性は、個人技からチーム設計へ
moongift
PRO
4
2.5k
Alphaモジュール使っていいのかい!?いけないのかい!?どっちなんだいっ!?
watany
1
320
ファミコンでPHPを動かす / PHP on the Famicom
tomzoh
2
610
2026年のソフトウェア開発を考える(2026/07版) / Agentic Software Engineering 2026-07 Findy Edition
twada
PRO
26
11k
AI Native なプロダクト組織の立ち上げ方 : 生産性 100 倍への挑戦
mikesorae
0
1.3k
Jitera Company Deck
jitera
0
280
AI時代のPlaywright活用(システムテストを自動化する ー 実行エンジンにPla ywrightを選んだ理由)
ynisqa1988
2
950
それでも、技術なブログを書く理由 #kichijojipm / Why I Still Write Tech Blogs Even Now
shinkufencer
0
450
複数プロダクト組織のAIネイティブ化における戦略 / AICon2026_kude
rakus_dev
0
300
Playwright × AI Agent でE2Eテストはどう変わるか AI駆動テストの可能性と実用検証の結果
taiga7543
2
810
Featured
See All Featured
JavaScript: Past, Present, and Future - NDC Porto 2020
reverentgeek
52
6k
Claude Code どこまでも/ Claude Code Everywhere
nwiizo
65
57k
A Tale of Four Properties
chriscoyier
163
24k
Speed Design
sergeychernyshev
33
1.9k
Optimizing for Happiness
mojombo
378
71k
Sharpening the Axe: The Primacy of Toolmaking
bcantrill
46
2.9k
Creating an realtime collaboration tool: Agile Flush - .NET Oxford
marcduiker
35
2.5k
CSS Pre-Processors: Stylus, Less & Sass
bermonpainter
360
30k
Music & Morning Musume
bryan
47
7.3k
Crafting Experiences
bethany
1
230
"I'm Feeling Lucky" - Building Great Search Experiences for Today's Users (#IAC19)
danielanewman
230
23k
How to optimise 3,500 product descriptions for ecommerce in one day using ChatGPT
katarinadahlin
PRO
1
3.7k
Transcript
【第5回】ゼロから始めるゲノム解析 (Python編) Computing GC Content: Parsing FASTA and Analyzing Sequences
@kimoton
本勉強会の概要・目的 書籍名 対象者/目的 Mastering Python for Bioinformatics Python・バイオインフォ知識ほぼゼロの人 を対象に、正しいPythonのコーディング手 法について学ぶ
頻度 毎週〜隔週開催予定 登壇者 募集中!
Rosalindとは • 問題解決を通じてバイオインフォマティク ス、プログラミング、およびアルゴリズムを 学習するためのプラットフォーム • 大学やハッカソン、就職の面接にも 600回 以上の採用実績あり 参考:https://qiita.com/_kimoton/items/d534d0fa9b83dd7dc412
概要
環境構築 - 必要パッケージ群のインストール # 公開されているレポジトリからファイル群を取得 $ git clone https://github.com/kyclark/biofx_python $
cd biofx_python # requirements.txt に記載のパッケージをインストール $ pip3 install -r requirements.txt # pylintの設定ファイルをホームディレクトリに移動 $ cp pylintrc ~/.pylintrc # mypyの設定ファイルをホームディレクトリに移動 $ cp mypy.ini ~/.mypy.ini
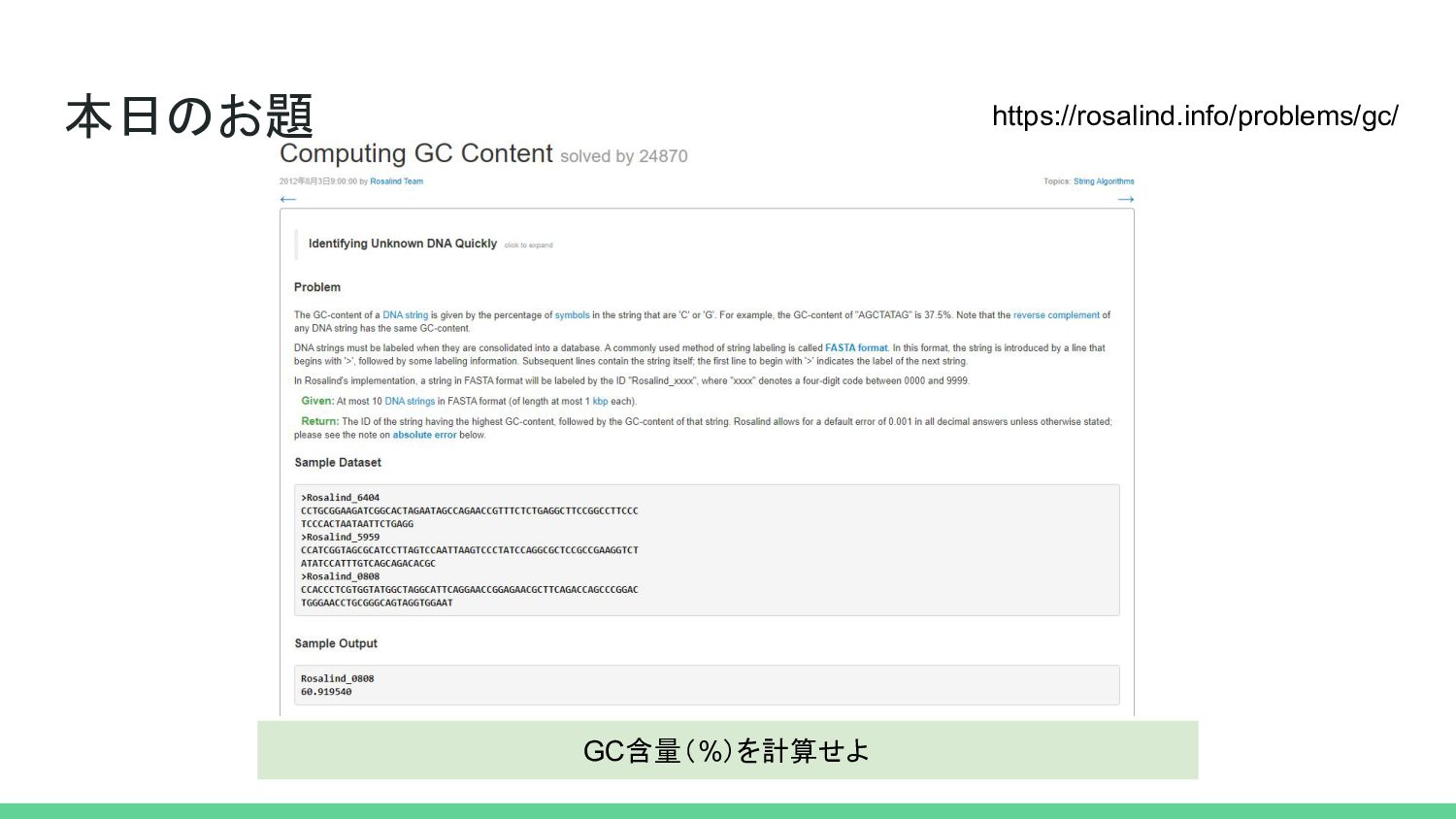
本日のお題 GC含量(%)を計算せよ https://rosalind.info/problems/gc/
本日学ぶこと • Bio.Seqモジュールを用いたFASTAファイルのパースの仕方 • 標準入力の受け取り方 • formatメソッドを用いた文字列フォーマットの仕方 • リスト内包表記、filter()関数、map()関数を用いたforループの書き換 え
• 正規表現を用いた文字列内のパターンカウント
前提知識編
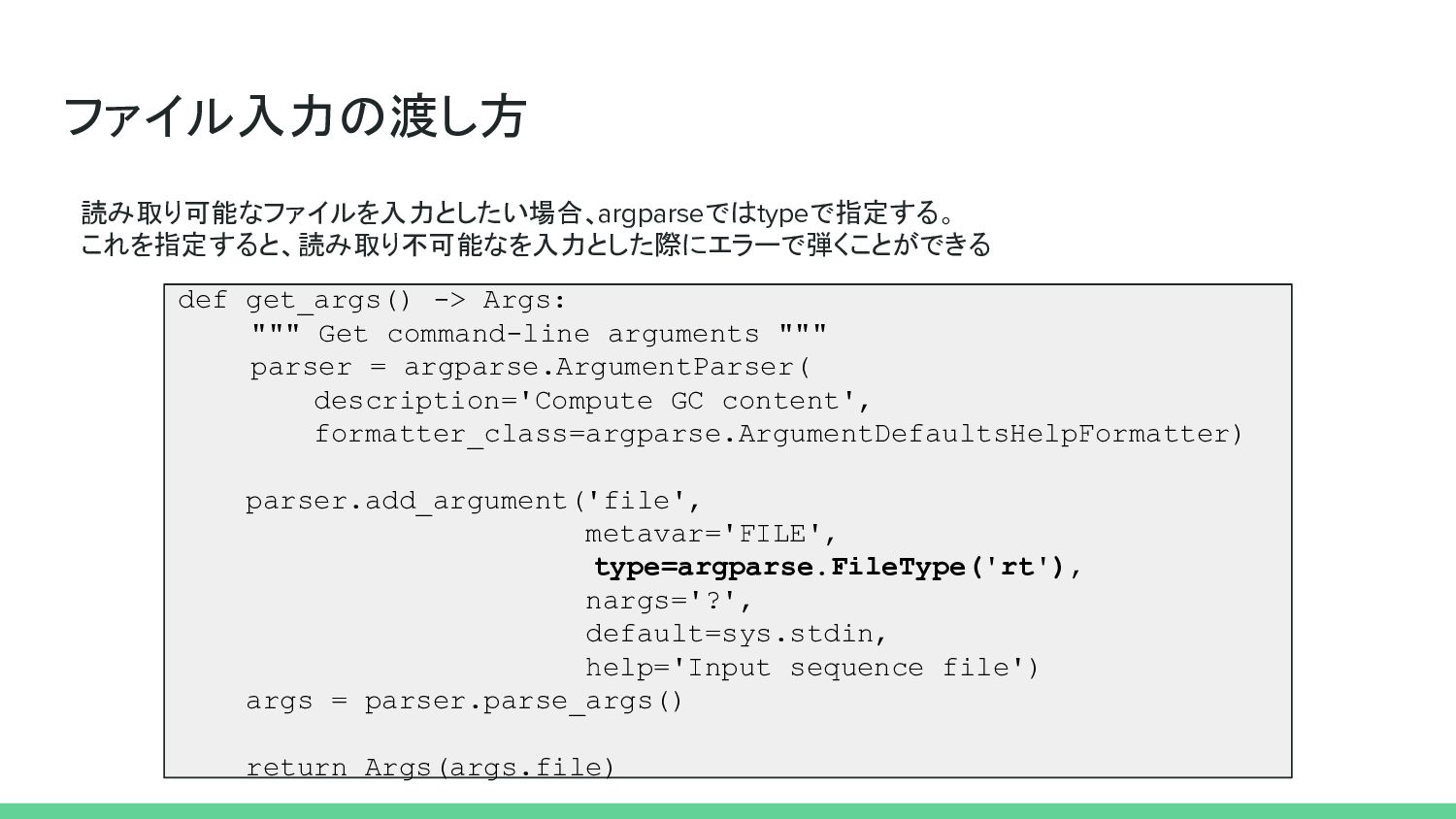
ファイル入力の渡し方 読み取り可能なファイルを入力としたい場合、argparseではtypeで指定する。 これを指定すると、読み取り不可能なを入力とした際にエラーで弾くことができる def get_args() -> Args: """ Get command-line
arguments """ parser = argparse.ArgumentParser( description='Compute GC content', formatter_class=argparse.ArgumentDefaultsHelpFormatter) parser.add_argument('file', metavar='FILE', type=argparse.FileType('rt'), nargs='?', default=sys.stdin, help='Input sequence file') args = parser.parse_args() return Args(args.file)
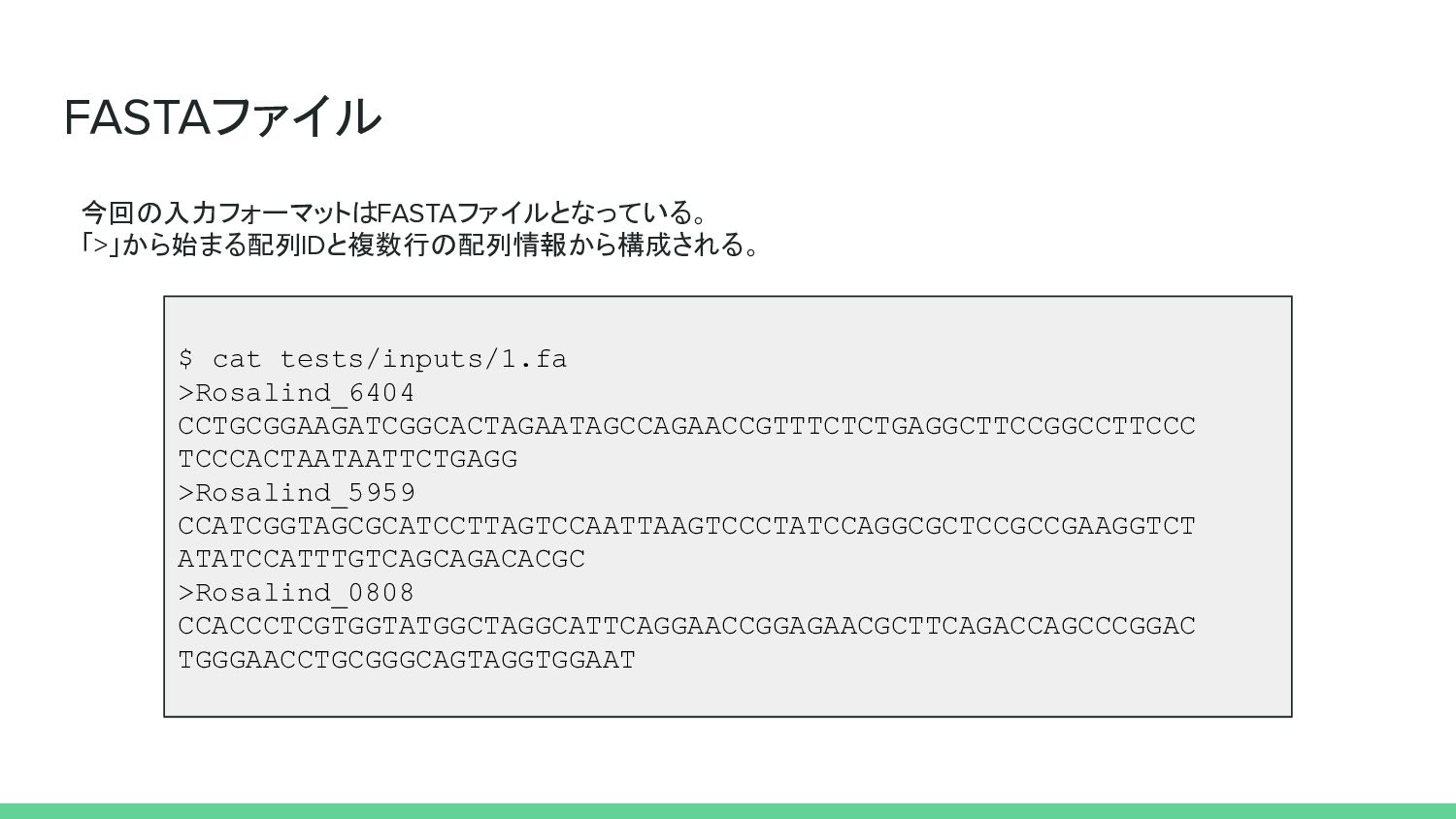
FASTAファイル 今回の入力フォーマットはFASTAファイルとなっている。 「>」から始まる配列IDと複数行の配列情報から構成される。 $ cat tests/inputs/1.fa >Rosalind_6404 CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGCCTTCCC TCCCACTAATAATTCTGAGG >Rosalind_5959
CCATCGGTAGCGCATCCTTAGTCCAATTAAGTCCCTATCCAGGCGCTCCGCCGAAGGTCT ATATCCATTTGTCAGCAGACACGC >Rosalind_0808 CCACCCTCGTGGTATGGCTAGGCATTCAGGAACCGGAGAACGCTTCAGACCAGCCCGGAC TGGGAACCTGCGGGCAGTAGGTGGAAT
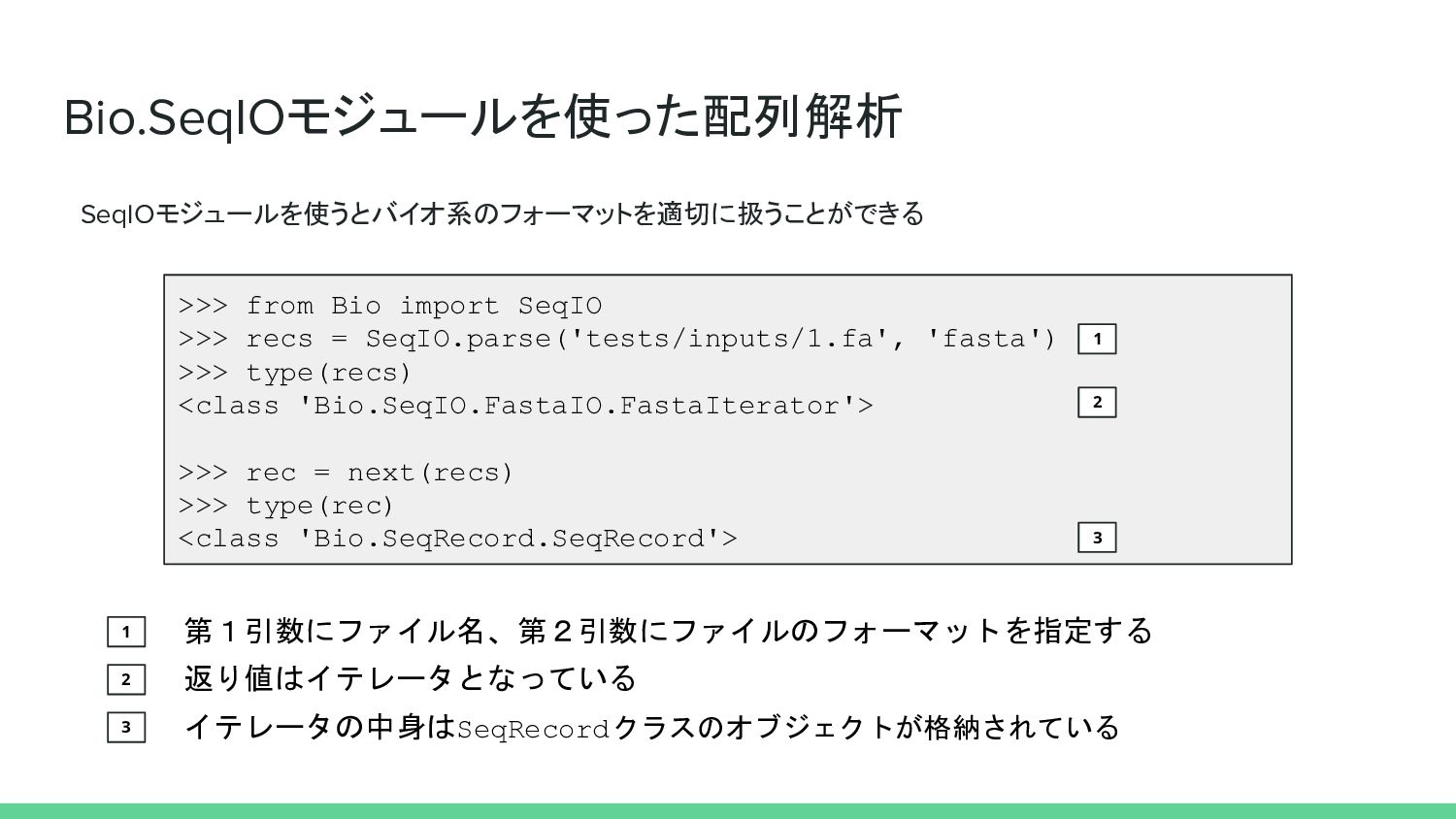
Bio.SeqIOモジュールを使った配列解析 SeqIOモジュールを使うとバイオ系のフォーマットを適切に扱うことができる >>> from Bio import SeqIO >>> recs =
SeqIO.parse('tests/inputs/1.fa', 'fasta') >>> type(recs) <class 'Bio.SeqIO.FastaIO.FastaIterator'> >>> rec = next(recs) >>> type(rec) <class 'Bio.SeqRecord.SeqRecord'> 1 1 第1引数にファイル名、第2引数にファイルのフォーマットを指定する 2 返り値はイテレータとなっている 2 3 3 イテレータの中身はSeqRecordクラスのオブジェクトが格納されている
SeqRecordクラス SeqIOモジュールで読み取ったデータはSeqRecordクラスのオブジェクトとして扱う >>> rec SeqRecord( seq=Seq('CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGC...AGG'), id='Rosalind_6404', name='Rosalind_6404', description='Rosalind_6404', dbxrefs=[]
) 1 1 seq属性には、FASTAファイルに含まれていた配列のSeqオブジェクトが格納され ている 2 id属性には、FASTAファイルに含まれていた配列IDが格納される 2 3 他にもnameやdescriptionといった情報が格納できる。 3
SeqRecordクラスからの配列情報の取り出し 配列情報は.seq属性に含まれている。 Seqオブジェクトとして扱うと配列に係わる様々な便利なモジュールが使える。 # 配列情報の取り出し >>> rec.seq Seq('CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGC...AGG') # 相補鎖配列を使用する場合は
reverse_complement メソッドを使う(第3回の内容) >>> rec.seq.reverse_complement() Seq('CCTCAGAATTATTAGTGGGAGGGAAGGCCGGAAGCCTCAGAGAAACGGTTCTGG...AGG') 参考:https://biopython.org/docs/1.75/api/Bio.Seq.html
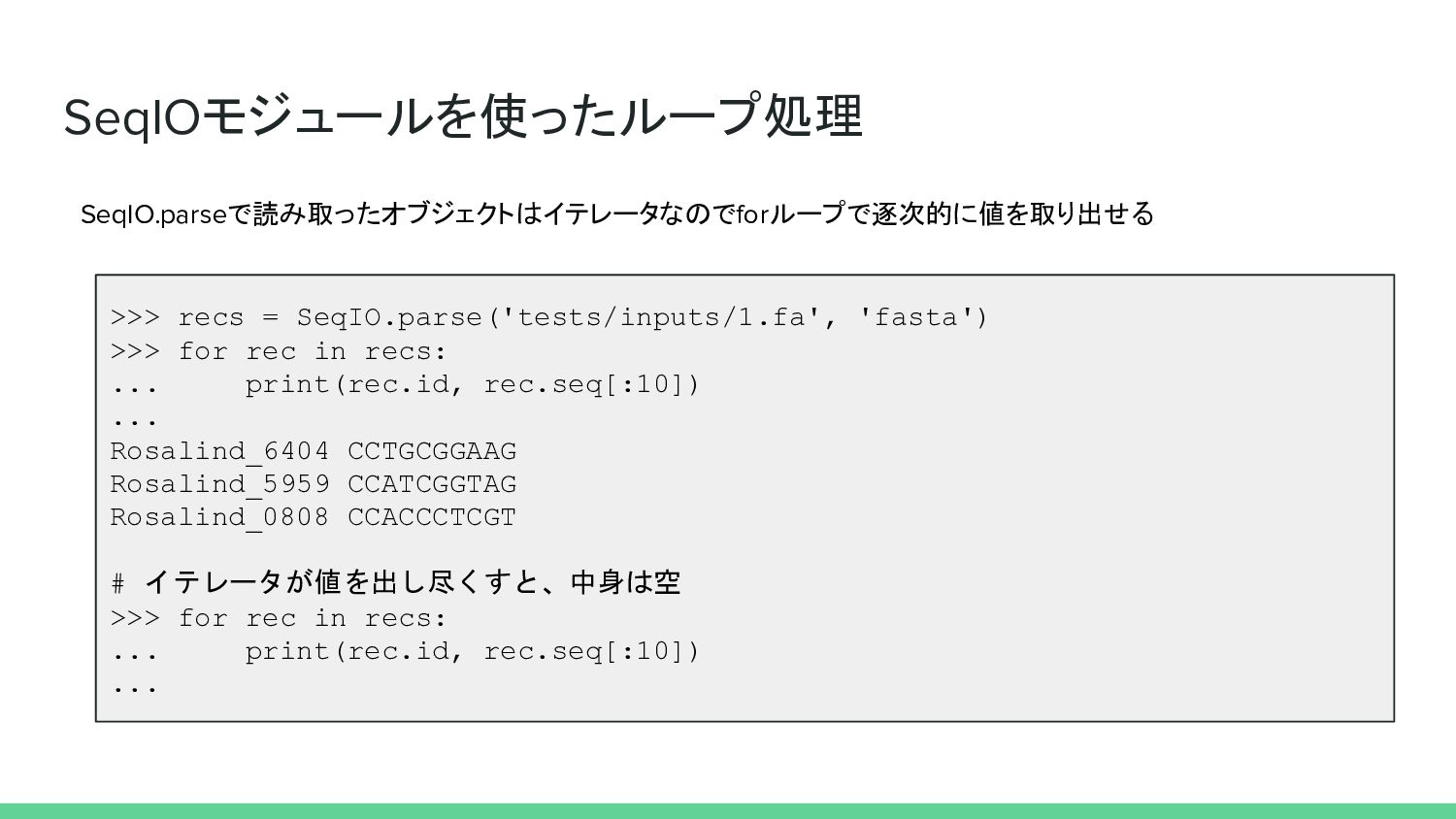
SeqIOモジュールを使ったループ処理 SeqIO.parseで読み取ったオブジェクトはイテレータなのでforループで逐次的に値を取り出せる >>> recs = SeqIO.parse('tests/inputs/1.fa', 'fasta') >>> for rec
in recs: ... print(rec.id, rec.seq[:10]) ... Rosalind_6404 CCTGCGGAAG Rosalind_5959 CCATCGGTAG Rosalind_0808 CCACCCTCGT # イテレータが値を出し尽くすと、中身は空 >>> for rec in recs: ... print(rec.id, rec.seq[:10]) ...
GC含量(個数)の算出 純粋にGC含量(個数)をカウントする場合以下のように実装すればよい seq = 'CCACCCTCGTGGTATGGCT' gc = 0 for base
in seq: if base in ('G', 'C'): gc += 1 1 1 GC含量を格納する変数を0で初期化 2 seqに格納された文字列 をイテレーション 3 ループしている文字列がGまたはCのいずれかであればカウントアップ 2 3
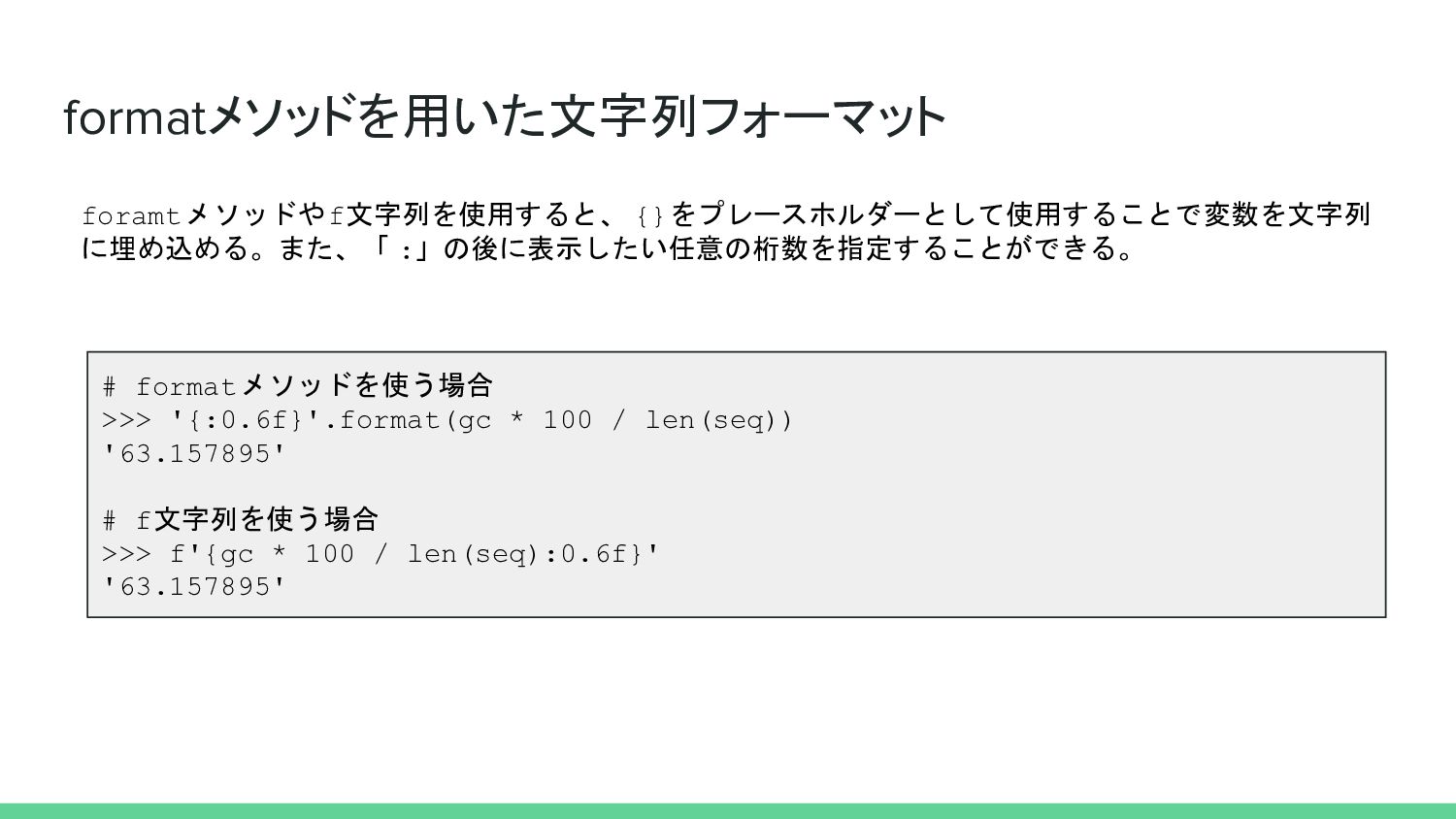
formatメソッドを用いた文字列フォーマット # formatメソッドを使う場合 >>> '{:0.6f}'.format(gc * 100 / len(seq)) '63.157895'
# f文字列を使う場合 >>> f'{gc * 100 / len(seq):0.6f}' '63.157895' foramtメソッドやf文字列を使用すると、 {}をプレースホルダーとして使用することで変数を文字列 に埋め込める。また、「 :」の後に表示したい任意の桁数を指定することができる。
解法編
解法1~3 demo 解法1 1. 配列IDごとにループし、GC含量とIDをセットにしたタプルを作成 2. 最大のGC含量を持つタプルを max()関数で取得し、f文字列で必要な情報 を出力 解法2
解法1を以下によって改善 1. 名前付きタプルを使用 2. テストを実装 解法3 解法2を以下によって改善(メモリ効率) 1. GC含量最大のデータのみを保持するようにループを回す
解法4~8 demo 解法4 ループをリスト内包表記に書き換え 解法5 ループをfilter()関数を使って書き換え 解法6 ループをmap()関数を使って書き換え 解法7 正規表現を使った検索の実装&処理の関数化
解法8 単体テストを実装
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![SeqRecordクラス SeqIOモジュールで読み取ったデータはSeqRecordクラスのオブジェクトとして扱う >>> rec SeqRecord( seq=Seq('CCTGCGGAAGATCGGCACTAGAATAGCCAGAACCGTTTCTCTGAGGCTTCCGGC...AGG'), id='Rosalind_6404', name='Rosalind_6404', description='Rosalind_6404', dbxrefs=[]](https://files.speakerdeck.com/presentations/e82256e8a4af4cd78ba1b961bca552aa/slide_10.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}