(Gaidatzis, Lerch, Hahne, et al. 2015) • Rsubread (Liao, Smyth, and Shi 2013) • systemPipeR (Backman and Girke 2016) アライメントを行うための様々なRパッケージがbioconductor上で公開されている。
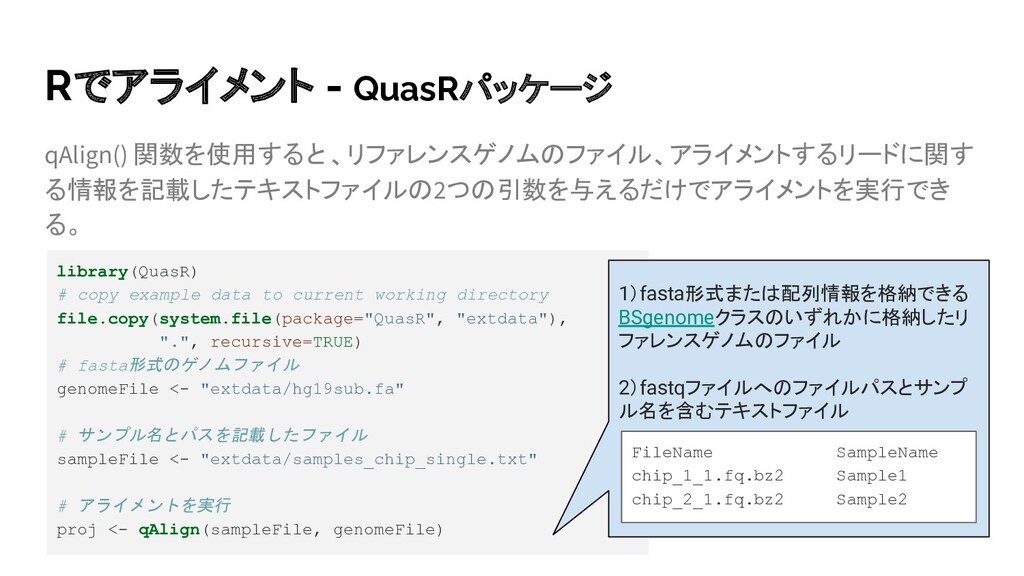
.fai file for: /home/kimoton/extdata/hg19sub.fa alignment files missing - need to: create alignment index for the genome create 2 genomic alignment(s) ・ ・ Genomic alignments have been created successfully # bamファイルが作成されていることを確認 > dir(pattern="\\.bam$", "extdata/") [1] "chip_1_1_3b0d53a66292.bam" "chip_2_1_3b0d3e161e85.bam" [3] "phiX_paired_withSecondary.bam" ゲノムインデックスが存在しな い場合は自動で生成。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}