(in Spanish) Introduction to the Python libraries argparse, SeqIO and NetworkX
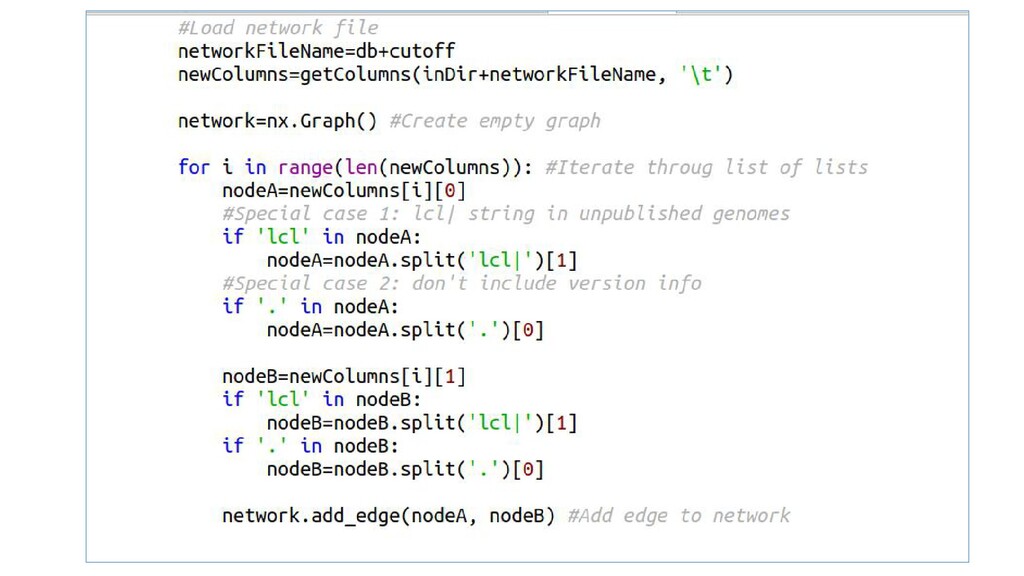
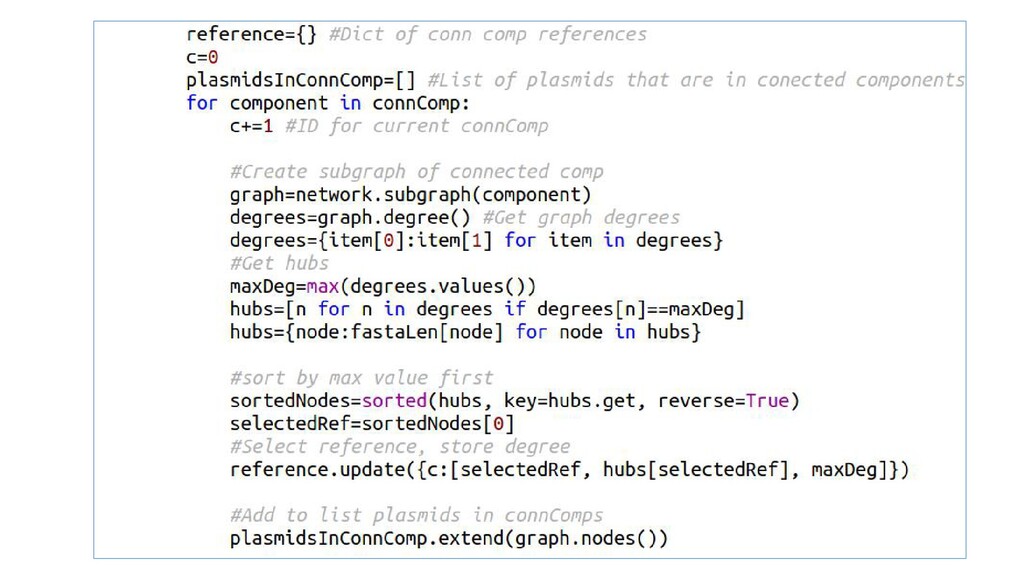
Examples with Python modules argparse for passing named command-line arguments, SeqIO to load and read biological data (genomic sequences), and NetworkX for the analysis of biological networks.
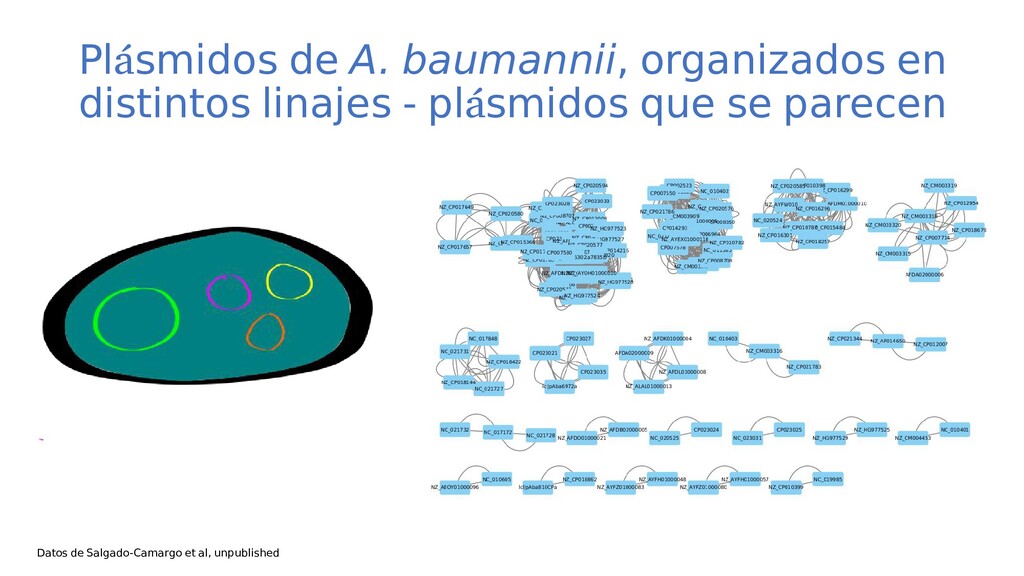
de bacterias problemáticas en los hospitales LCG Ma. Semiramis Castro Jaimes Programa de Doctorado en Ciencias Biomédicas Centro de Ciencias Genómicas, UNAM
https://www.illumina.com/content/dam/illumina-marketing/images/systems/v2/systems-carousel/system-carousel-miseq-front.png Photo by Hans-Peter Gauster on Unsplash: https://images.unsplash.com/photo-1494059980473-813e73ee784b?ixlib=rb-1.2.1&ixid=eyJhcHBfaWQiOjEyMDd9&auto=format&fit=crop&w=500&q=60
parámetros => muchos ensambles posibles organizados en carpetas •¿Es del tamaño esperado de un genoma del org. de interés? •¿Qué tan fragmentado está? •¿Tengo pedazos grandes?
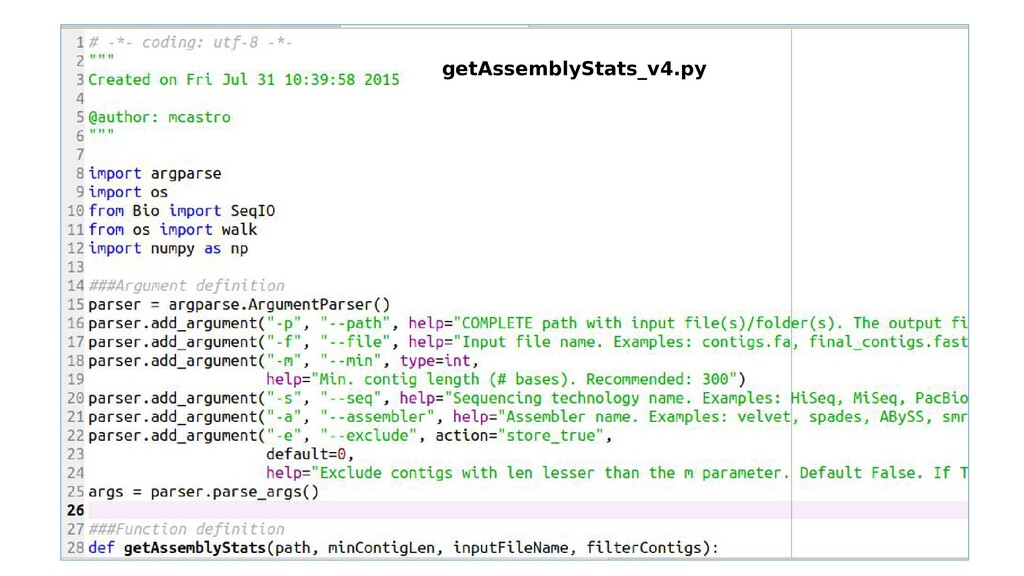
FILE] [-m MIN] [-s SEQ] [-a ASSEMBLER] [-e EXCLUDE] optional arguments: -h, --help show this help message and exit -p PATH, --path PATH COMPLETE path with input file(s)/folder(s). The output file will be written there. BioPython and Numpy must be installed -f FILE, --file FILE Input file name. Examples: contigs.fa, final_contigs.fasta, polished_assembly.fasta -m MIN, --min MIN Min. contig length (# bases). Recommended: 300 -s SEQ, --seq SEQ Sequencing technology name. Examples: HiSeq, MiSeq,PacBio, etc -a ASSEMBLER, --assembler ASSEMBLER Assembler name. Examples: velvet, spades, ABySS, smrtools, other -e EXCLUDE, --exclude EXCLUDE Exclude contigs with len lesser than the m parameter. Default False. If True, it will write a new file with prefix minLen_
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![argrparse: opciones y documentación usage: getAssemblyStats_v4.py [-h] [-p PATH] [-f](https://files.speakerdeck.com/presentations/e848134a49f54403b5c6e9d782862fad/slide_4.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}