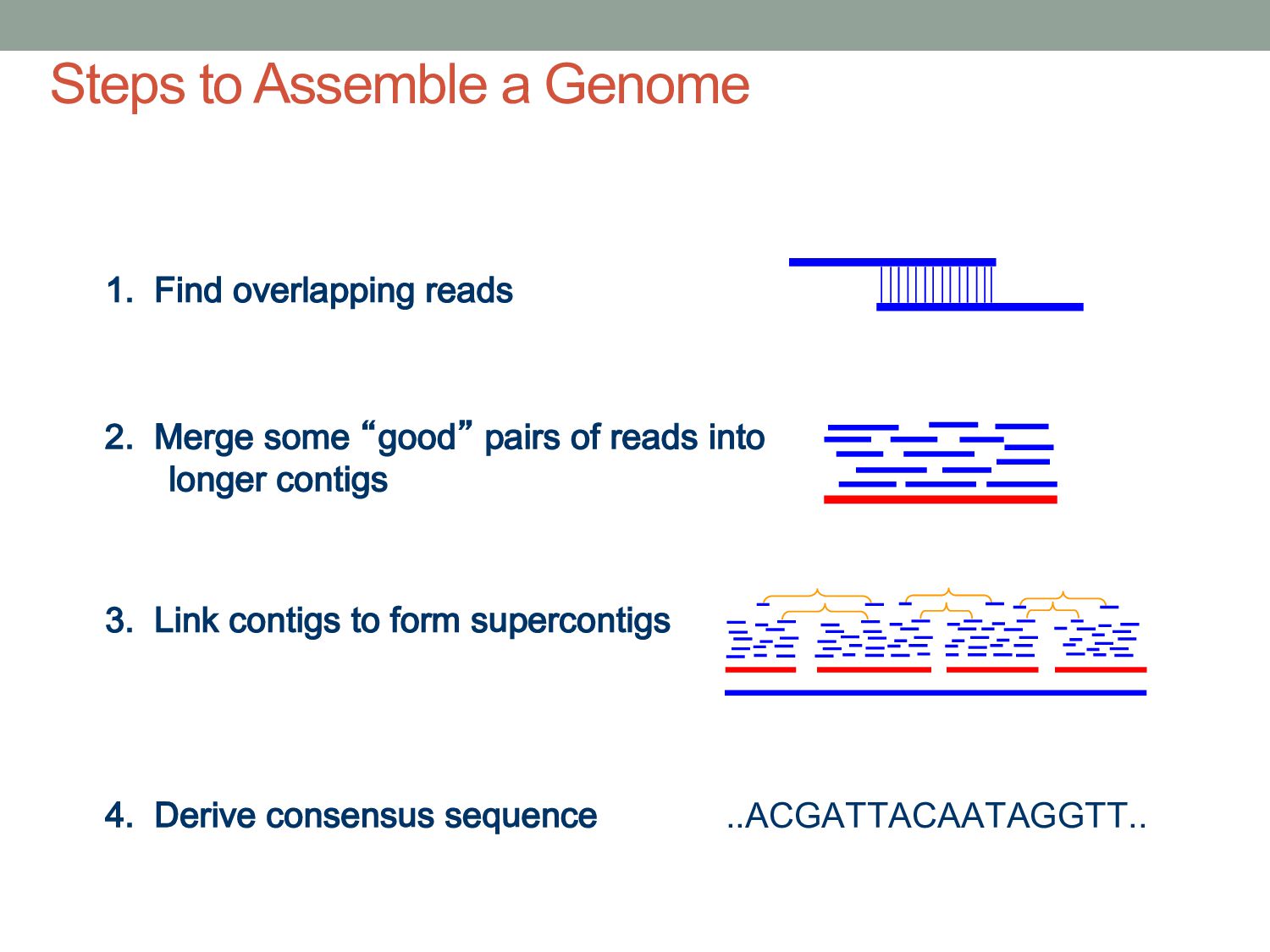
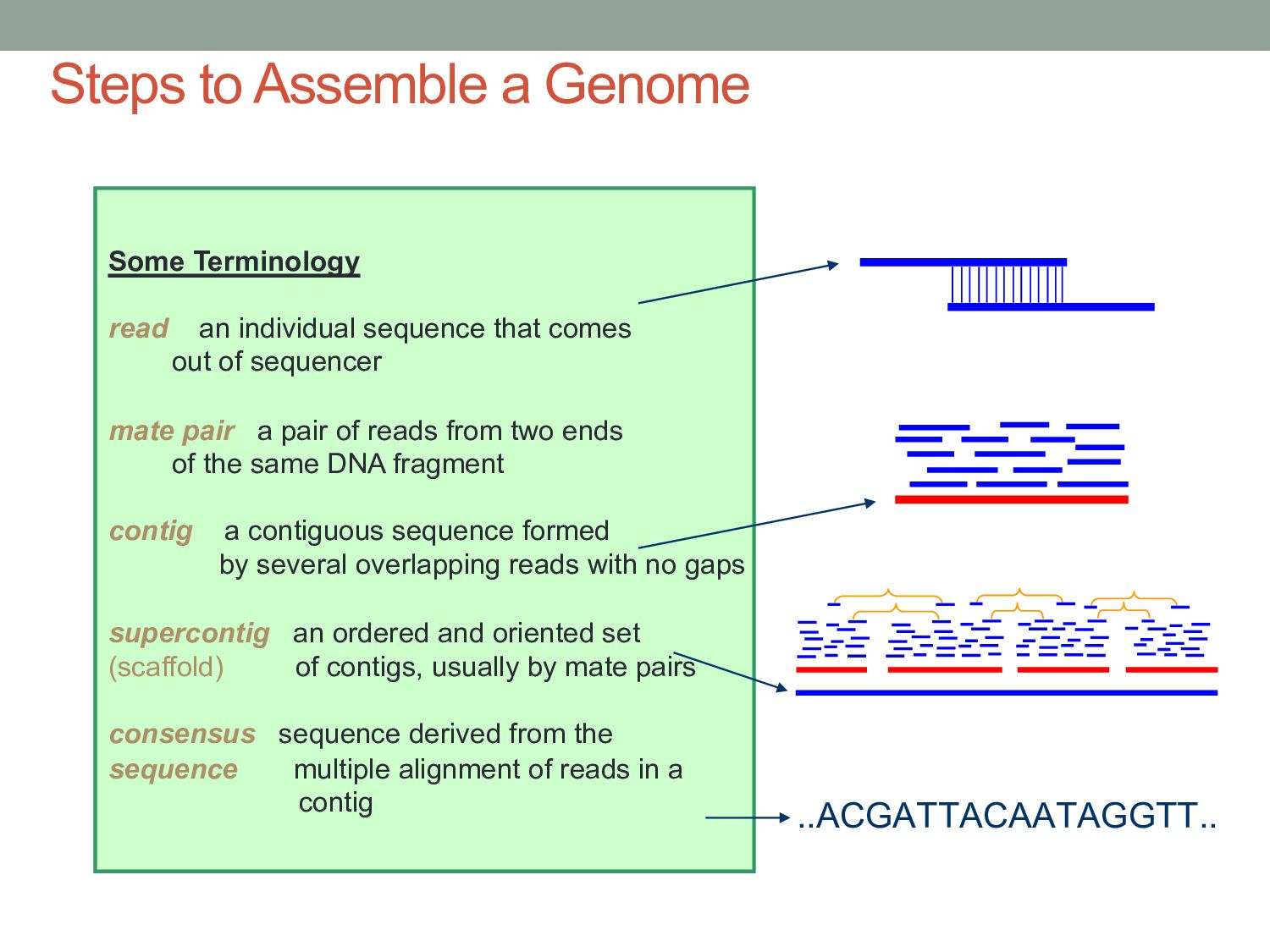
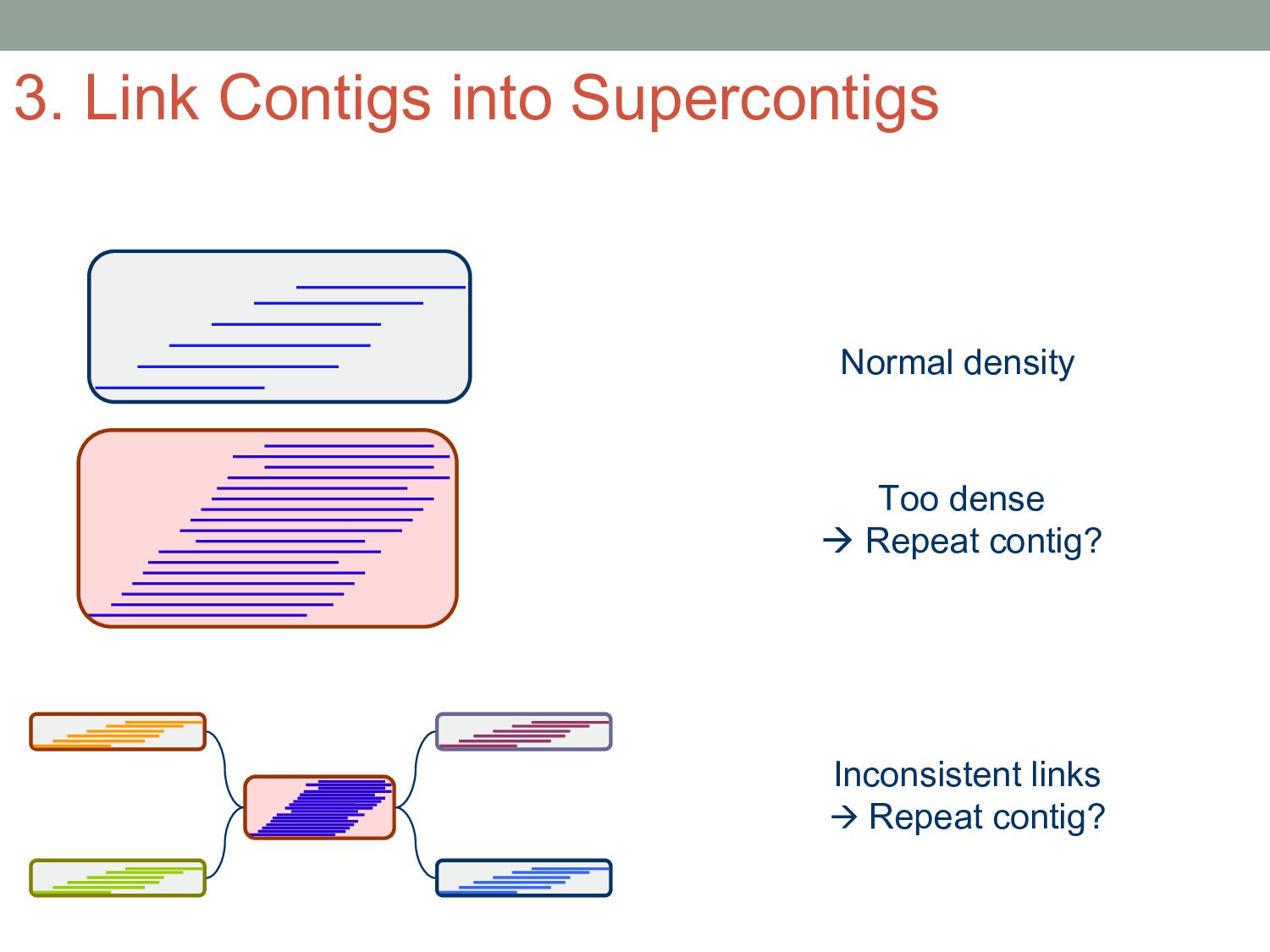
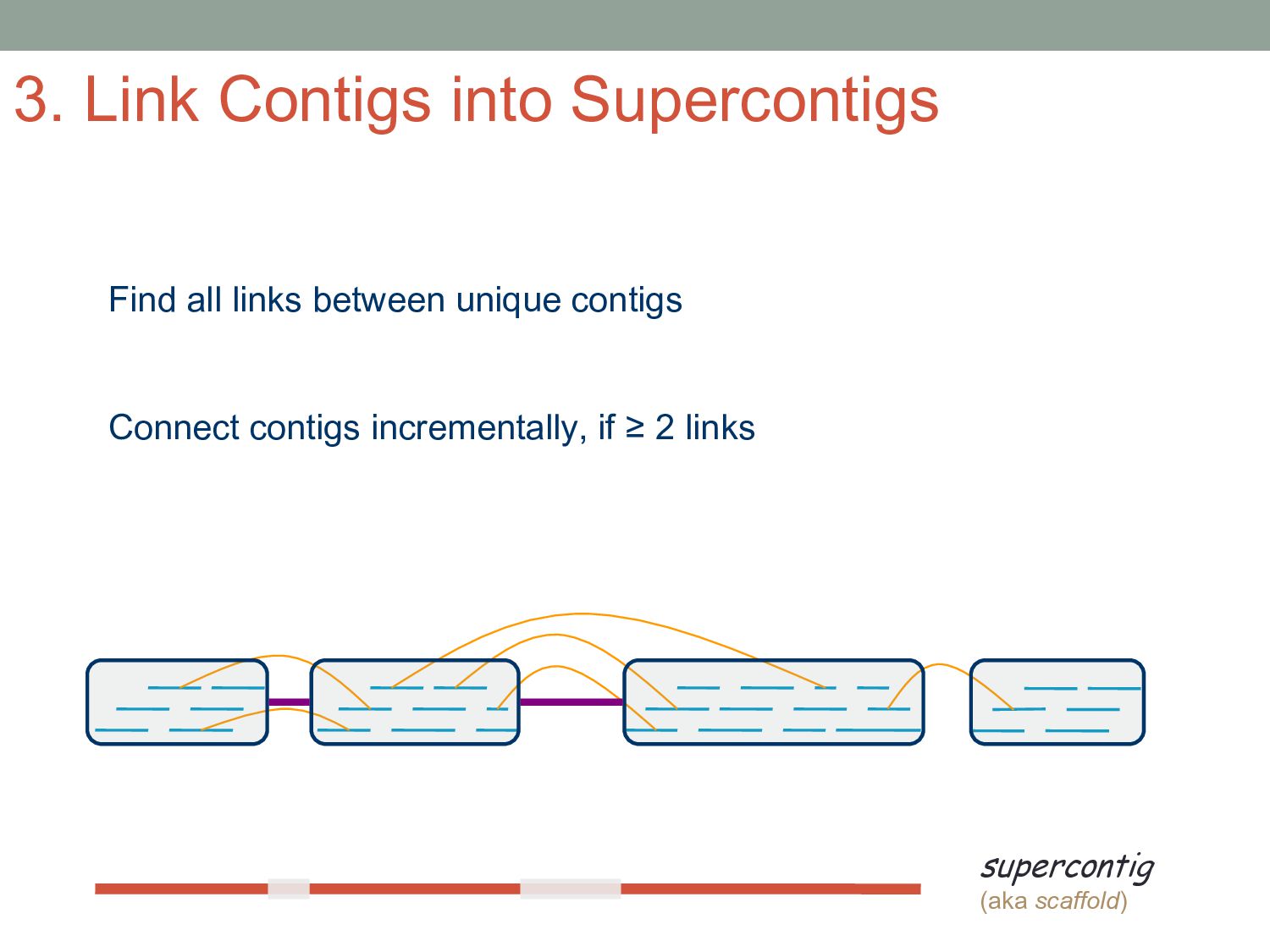
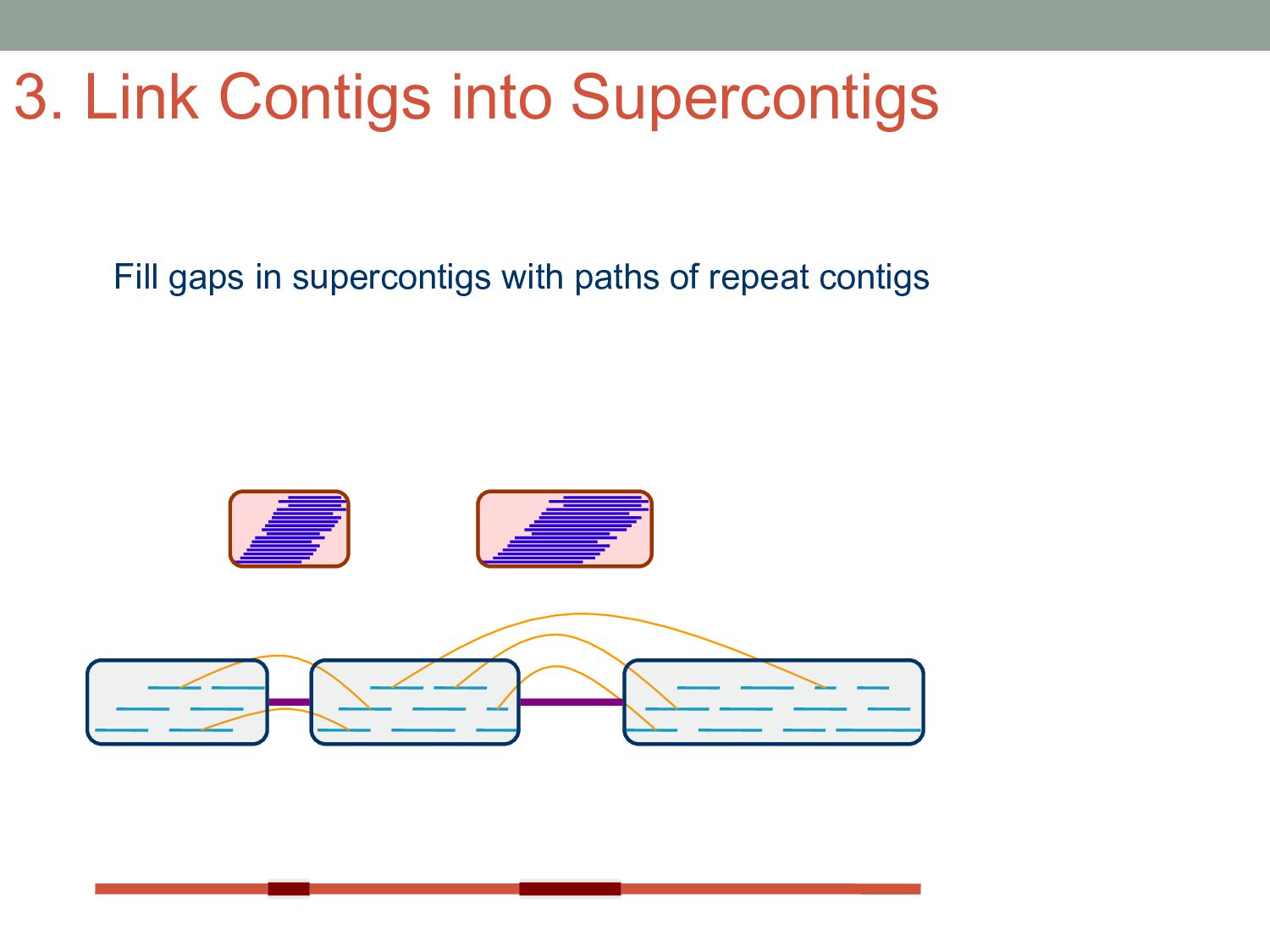
sequencer mate pair a pair of reads from two ends of the same DNA fragment contig a contiguous sequence formed by several overlapping reads with no gaps supercontig an ordered and oriented set (scaffold) of contigs, usually by mate pairs consensus sequence derived from the sequence multiple alignment of reads in a contig Steps to Assemble a Genome 77>?4>@@>?>>@>44@@77
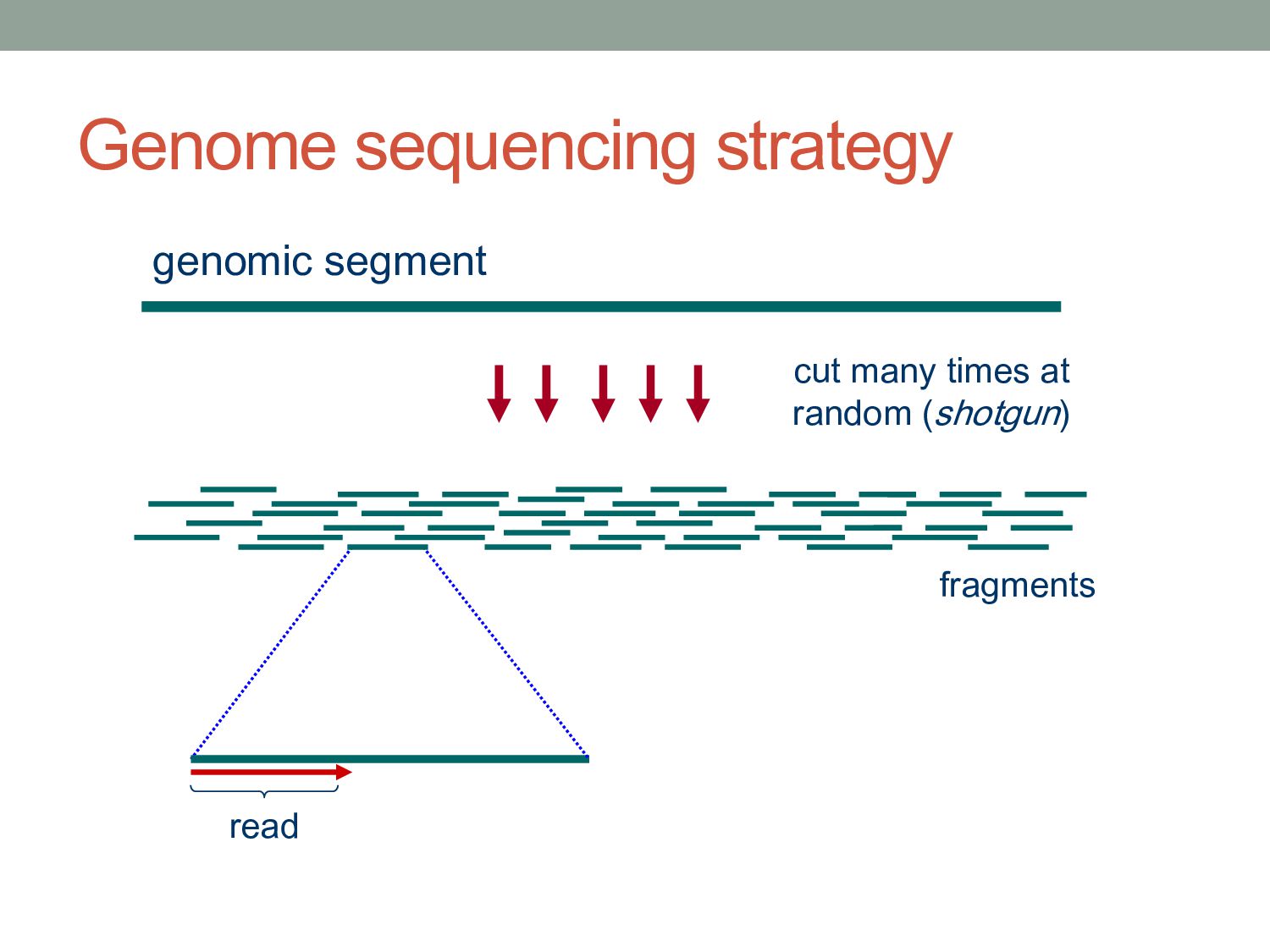
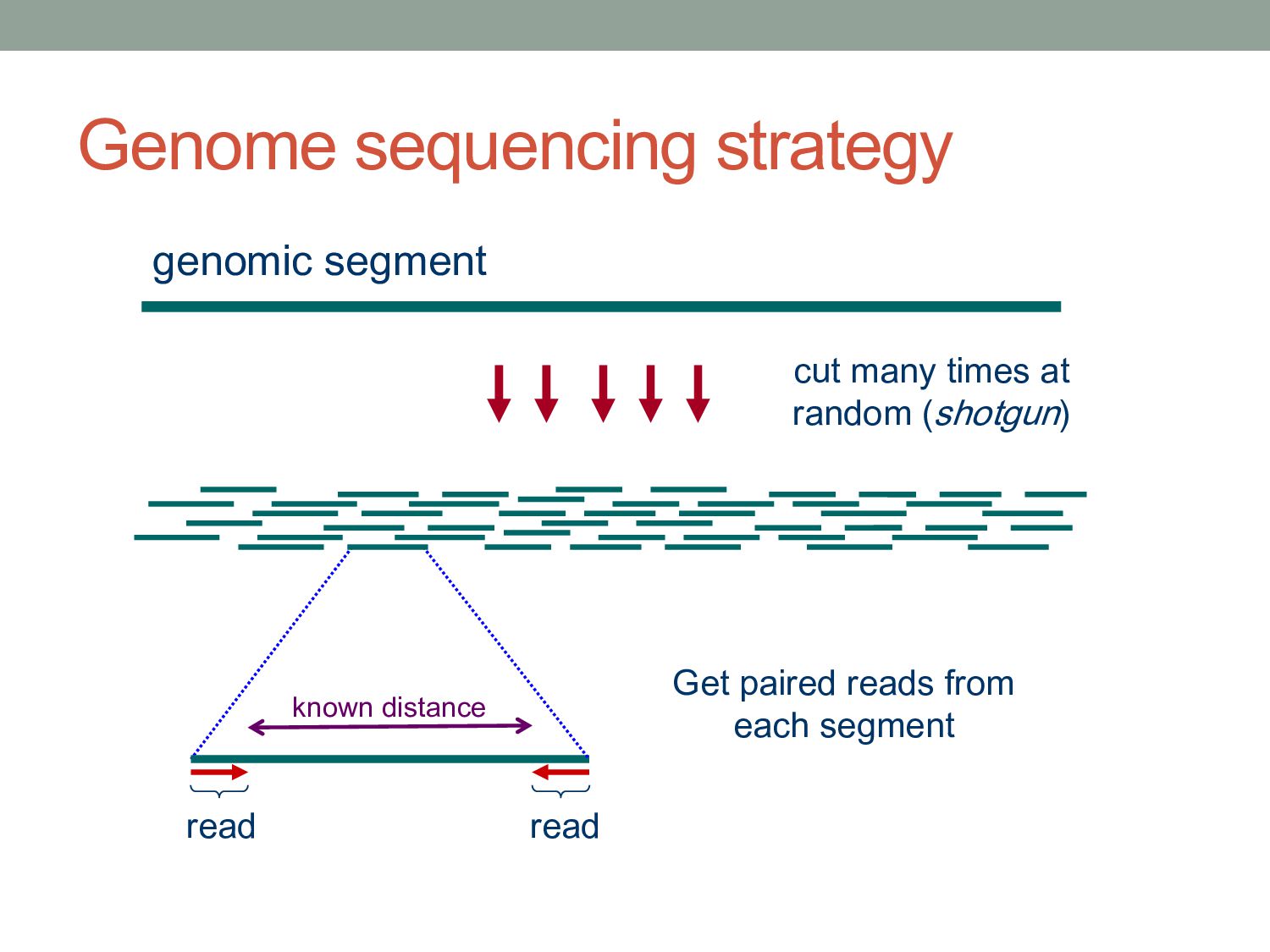
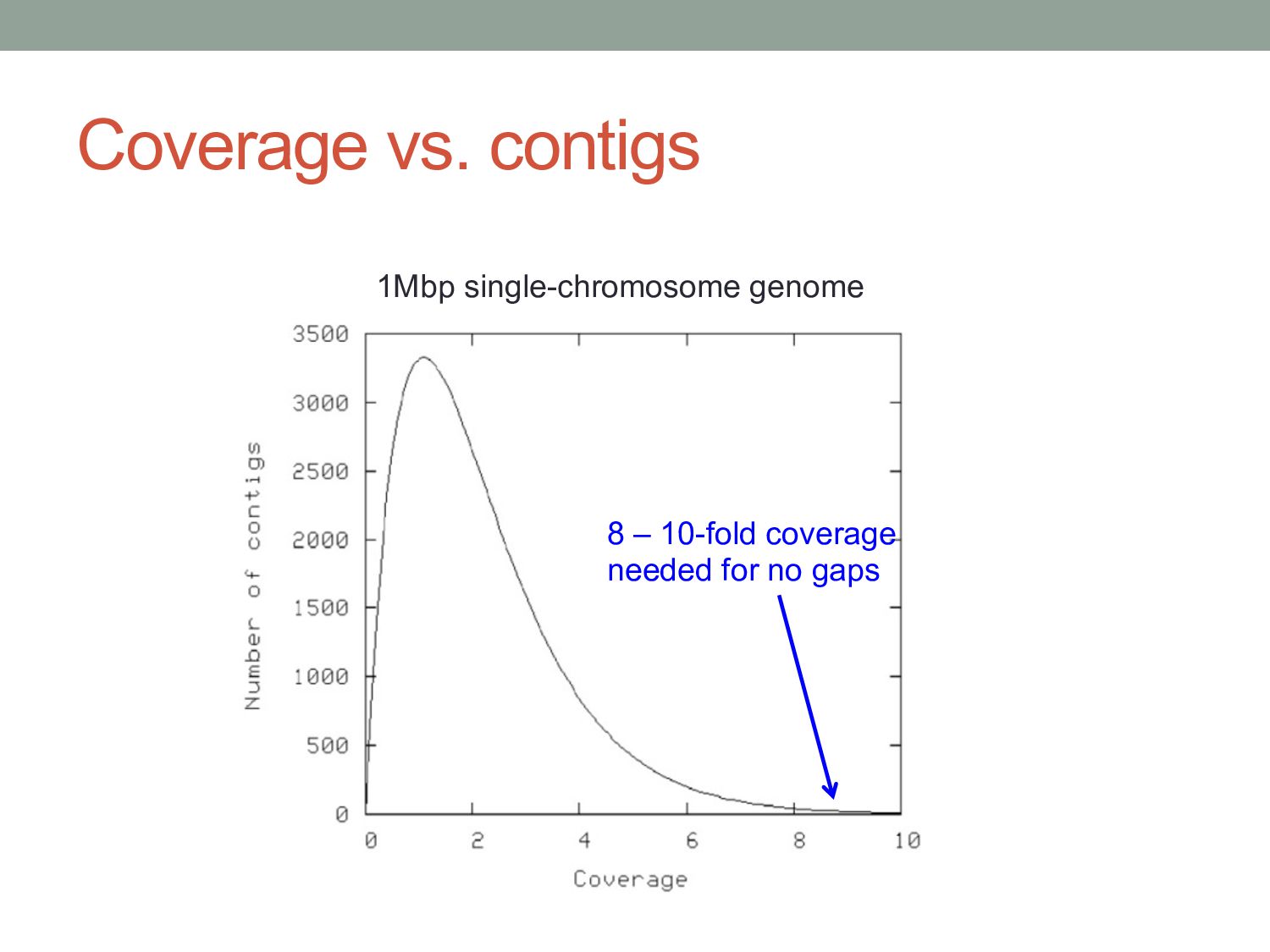
reads L = length of each read C = NL/G = “coverage” The expected number of gaps is given by: How many reads do we need to cover the whole genome? E(# gaps) = GC L e−C = Ne−C Described by a Poisson probability distribution
reads L = length of each read C = NL/G = “coverage” T = minimum detectable overlap between reads The expected number of gaps is given by: where α = 1 – T/L How many reads do we need to cover the whole genome? E(# gaps) = Ne−Cα
0 F1…i, 0 = 0 for each i = 1…M for each j = 1…N Fi-1, j-1 + s(Xi , Yj ) [match] Fi, j = max Fi-1, j – d [gap in X] Fi, j-1 – d [gap in Y] DIAG, if [match] Ptri, j = LEFT, if [gap in X] UP, if [gap in Y] Initialization Iteration Termination: Optimal alignment score is the maximum score in F{1…M},N or FM,{1…N}
a k-mer, k ~ 24 • Extend to full alignment F throw away if not >98% similar TAGATTACACAGATTAC TAGATTACACAGATTAC ||||||||||||||||| T GA TAGA | || TACA TAGT || • Caveat: repeats § A k-mer that occurs N times, causes O(N2) read/read comparisons § ALU k-mers could cause up to 1,000,0002 comparisons • Solution: § Discard all k-mers that occur !too often" • Set cutoff to balance sensitivity/speed tradeoff, according to genome at hand and computing resources available
TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAGATTACACAGATTATTGA TAGATTACACAGATTACTGA TAG-TTACACAGATTACTGA insert A replace T with C correlated errors— probably caused by repeats à disentangle overlaps TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAG-TTACACAGATTATTGA TAGATTACACAGATTACTGA TAG-TTACACAGATTATTGA In practice, error correction removes up to 98% of the errors TAGATTACACAGATTACTGA TAGATTACACAGATTACTGA TAG-TTACACAGATTATTGA TAGATTACACAGATTACTGA TAG-TTACACAGATTATTGA
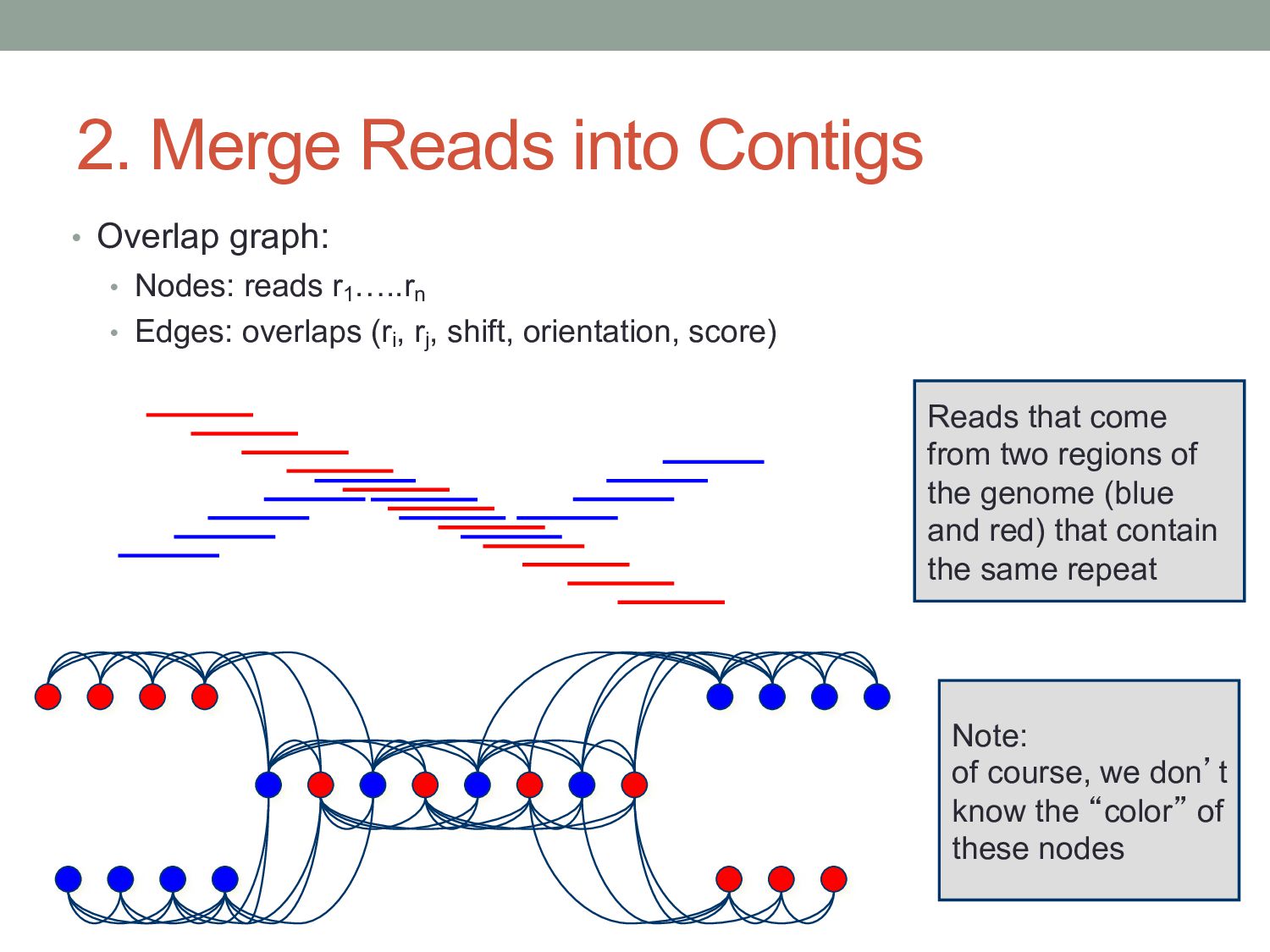
reads r1 …..rn • Edges: overlaps (ri , rj , shift, orientation, score) Note: of course, we don’t know the “color” of these nodes Reads that come from two regions of the genome (blue and red) that contain the same repeat
Green, U. Washington) • Method for calling base letters from Sanger sequencers • Phred quality scores are a convenient way to represent confidence in individual base calls. A C G A A T C A G 16 18 21 23 25 15 28 30 32 Quality scores: -10 * log10 (P(Error)) Phred score of 40: 10-40/10 chance of error = 99.99% confidence in call Phred score of 20: 10-20/10 chance of error = 99% confidence in call
Early assembler, widely used, good model of read errors • Overlap O(n2) à layout (no mate pairs) à consensus • Celera (Myers) • First assembler to handle large genomes (fly, human, mouse) • Overlap à layout à consensus • Arachne (Batzoglou) • Public assembler (mouse, several fungi) • Overlap à layout à consensus • Euler (Pevzner) • Indexing à de Bruijn graph à picking paths à consensus • Velvet (Birney) • Short reads à small genomes à simplification à error correction Overlapà layout à consensus String graphs, de Bruijn graphs
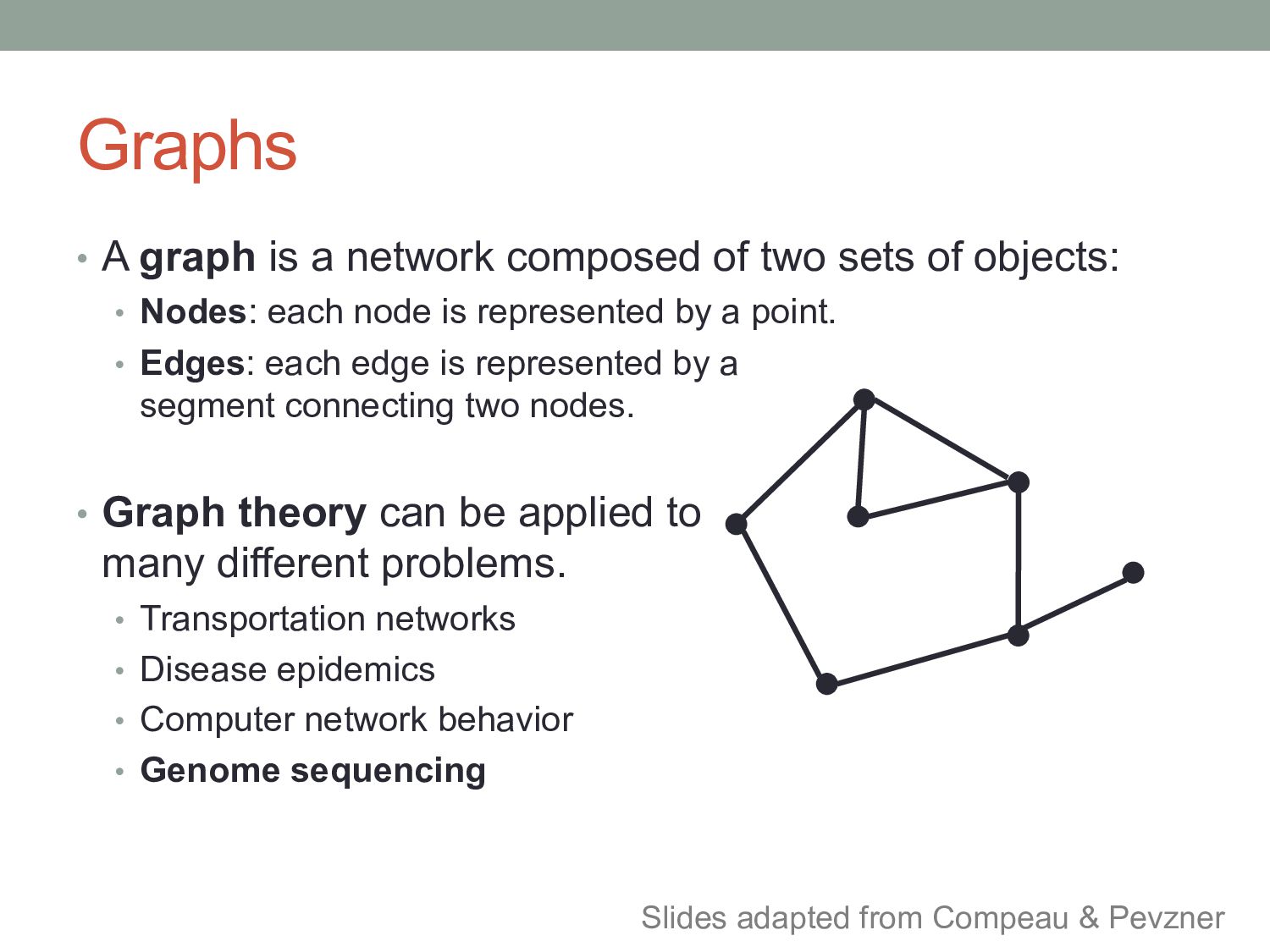
sets of objects: • Nodes: each node is represented by a point. • Edges: each edge is represented by a segment connecting two nodes. • Graph theory can be applied to many different problems. • Transportation networks • Disease epidemics • Computer network behavior • Genome sequencing Slides adapted from Compeau & Pevzner
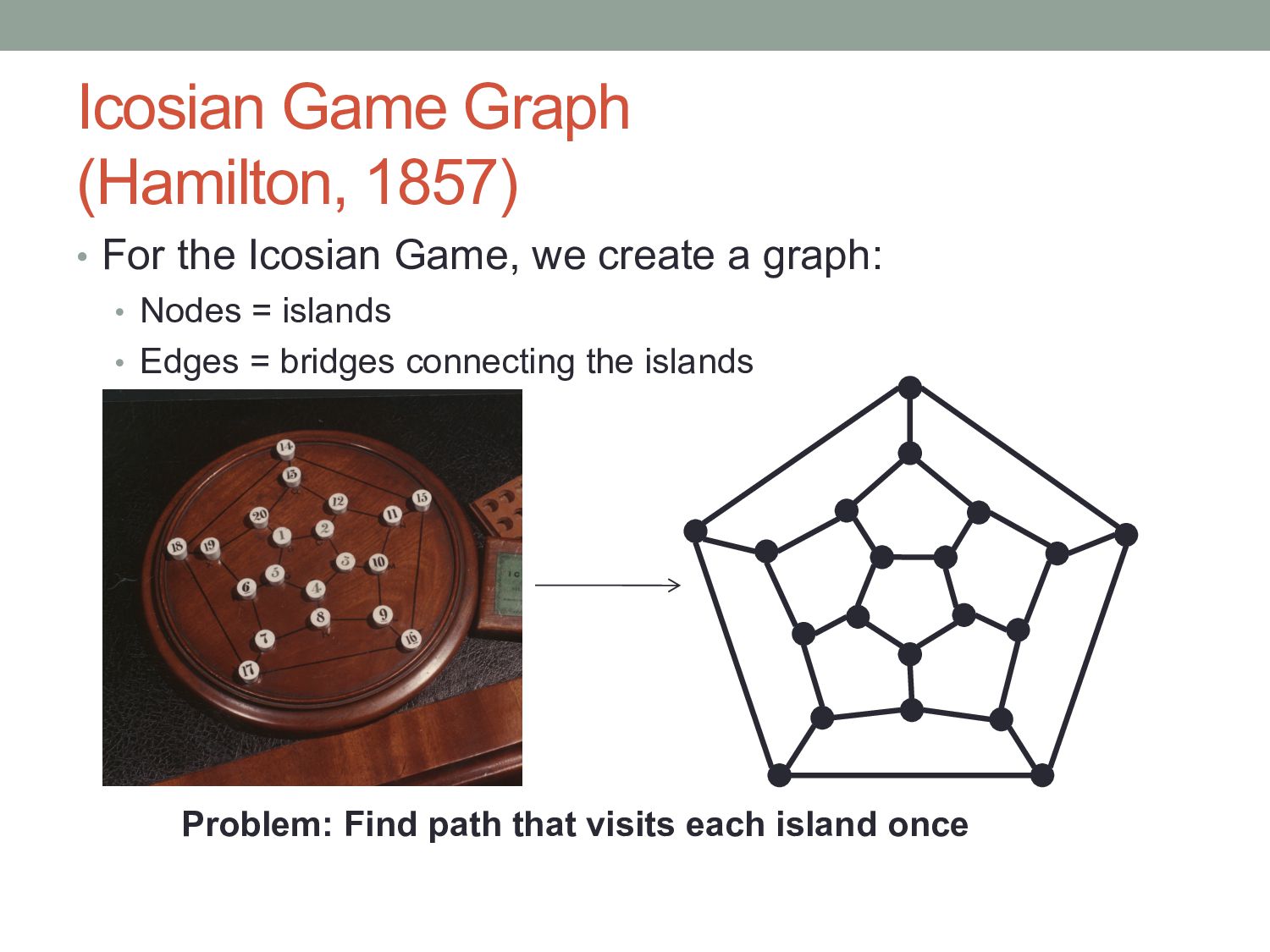
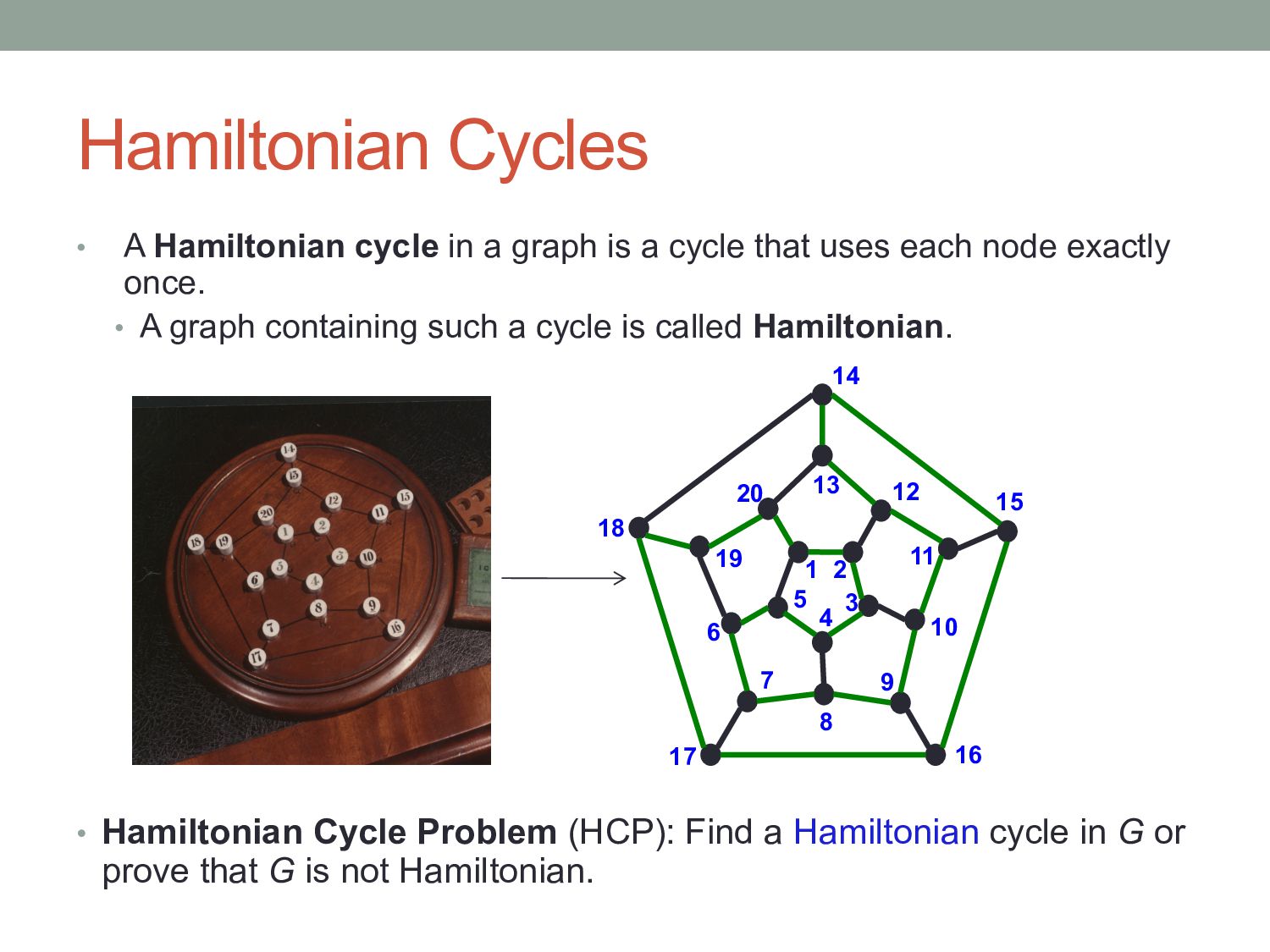
a cycle that uses each node exactly once. • A graph containing such a cycle is called Hamiltonian. • Hamiltonian Cycle Problem (HCP): Find a Hamiltonian cycle in G or prove that G is not Hamiltonian. 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
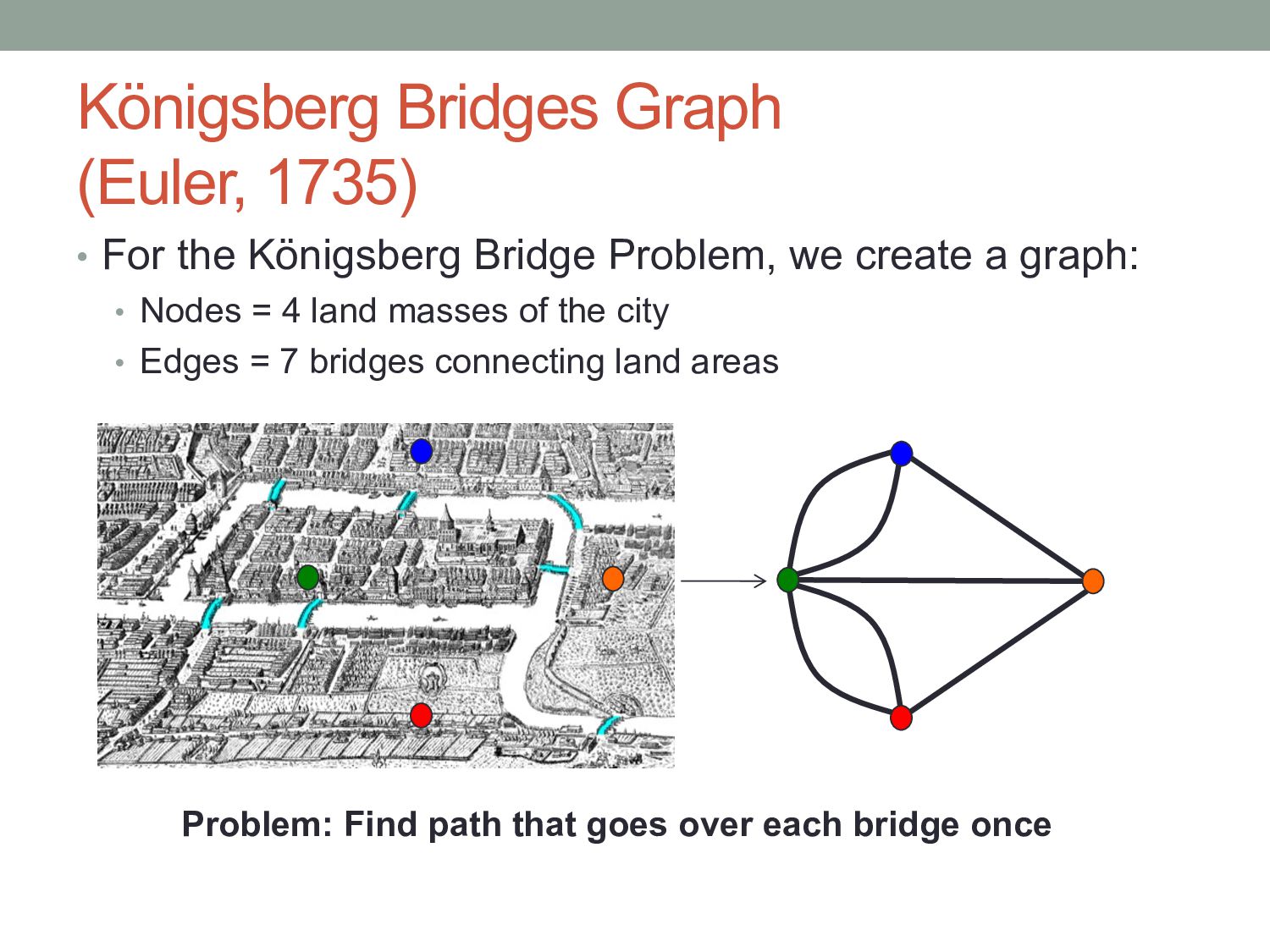
Problem, we create a graph: • Nodes = 4 land masses of the city • Edges = 7 bridges connecting land areas Problem: Find path that goes over each bridge once
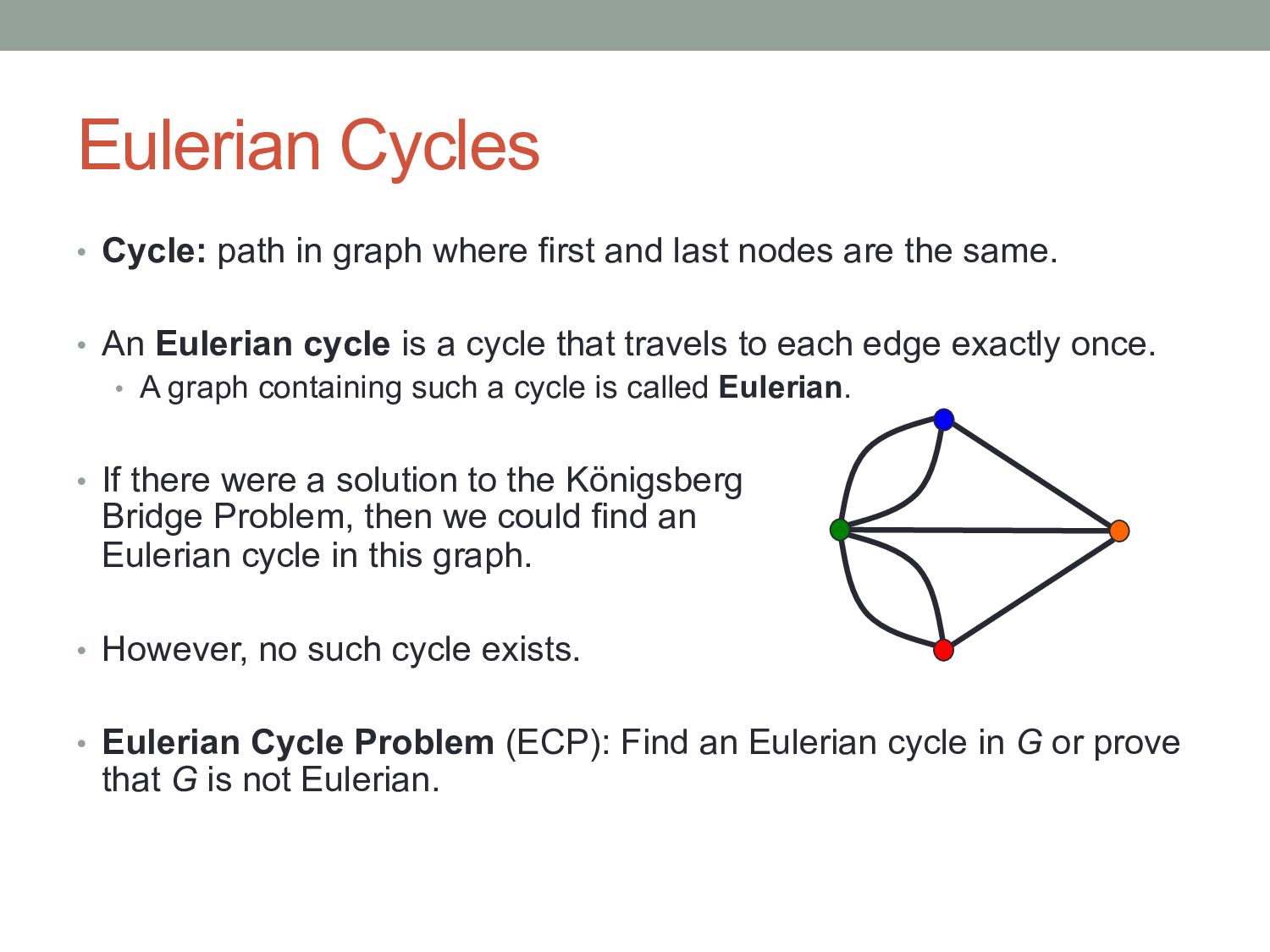
last nodes are the same. • An Eulerian cycle is a cycle that travels to each edge exactly once. • A graph containing such a cycle is called Eulerian. • If there were a solution to the Königsberg Bridge Problem, then we could find an Eulerian cycle in this graph. • However, no such cycle exists. • Eulerian Cycle Problem (ECP): Find an Eulerian cycle in G or prove that G is not Eulerian.
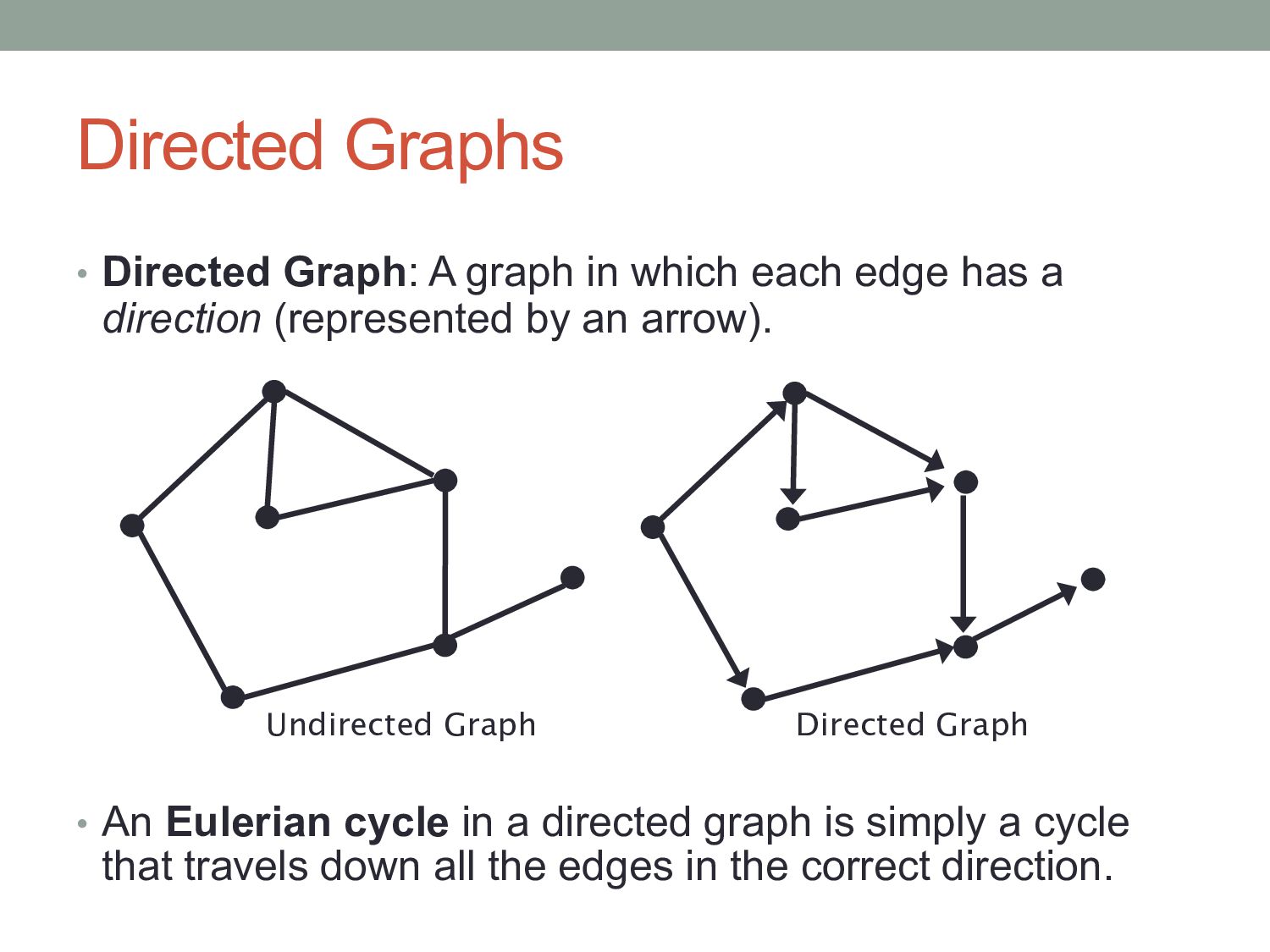
edge has a direction (represented by an arrow). • An Eulerian cycle in a directed graph is simply a cycle that travels down all the edges in the correct direction. Undirected Graph Directed Graph
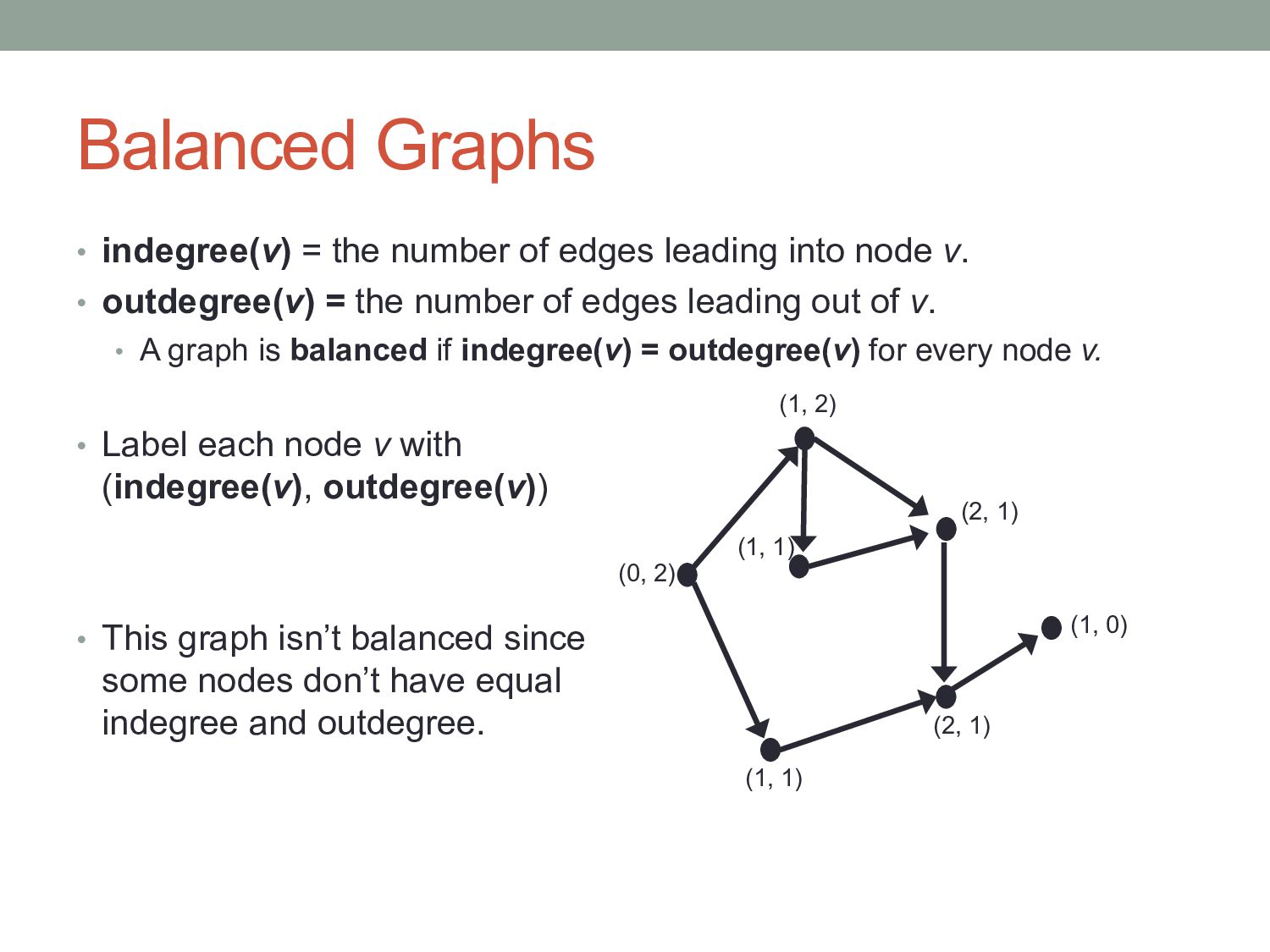
v. • outdegree(v) = the number of edges leading out of v. • A graph is balanced if indegree(v) = outdegree(v) for every node v. • Label each node v with (indegree(v), outdegree(v)) • This graph isn’t balanced since some nodes don’t have equal indegree and outdegree. Balanced Graphs (1, 2) (2, 1) (1, 0) (2, 1) (1, 1) (0, 2) (1, 1)
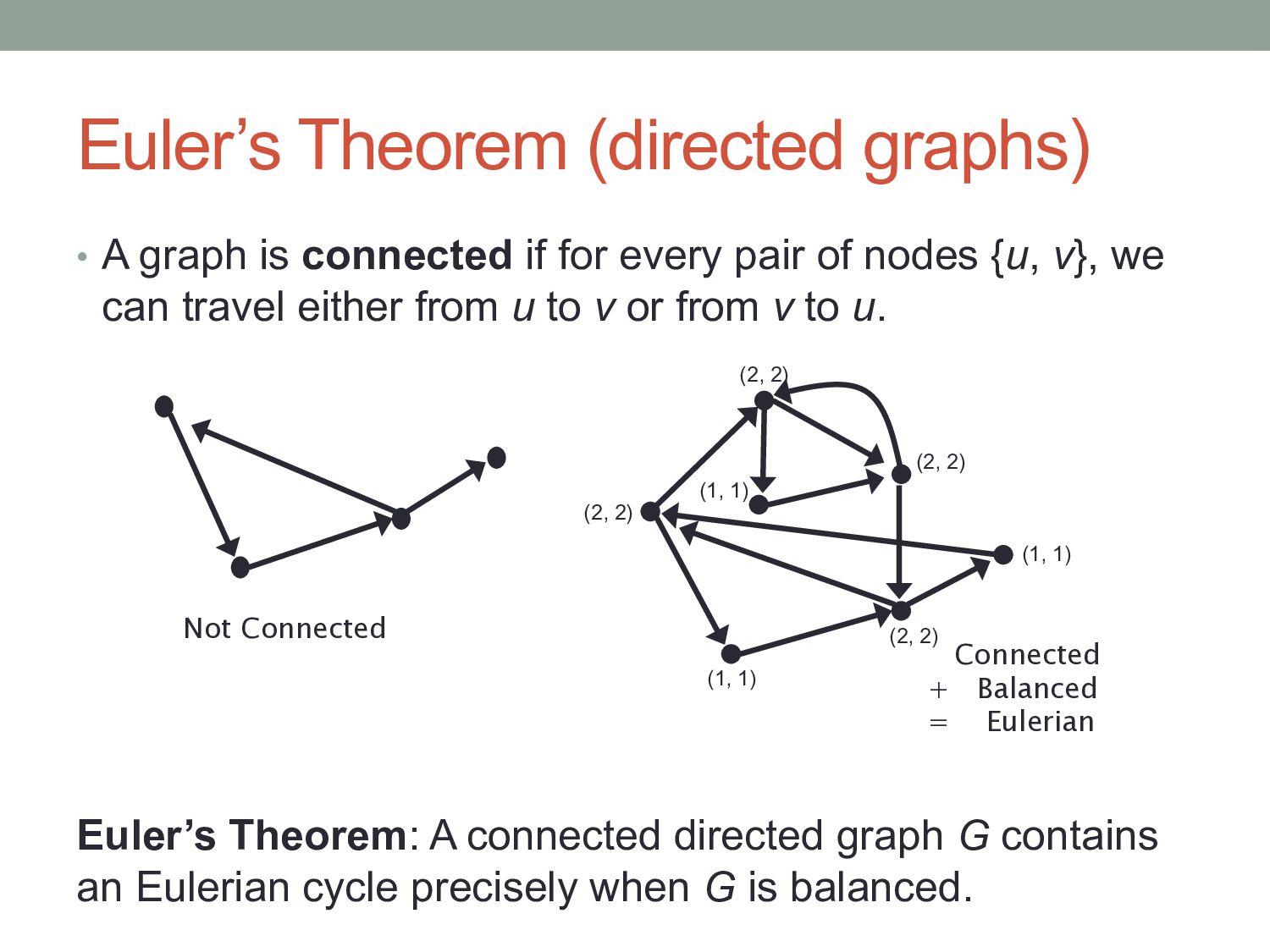
for every pair of nodes {u, v}, we can travel either from u to v or from v to u. Not Connected (2, 2) (2, 2) (1, 1) (2, 2) (1, 1) (2, 2) (1, 1) Connected + Balanced = Eulerian Euler’s Theorem: A connected directed graph G contains an Eulerian cycle precisely when G is balanced.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}