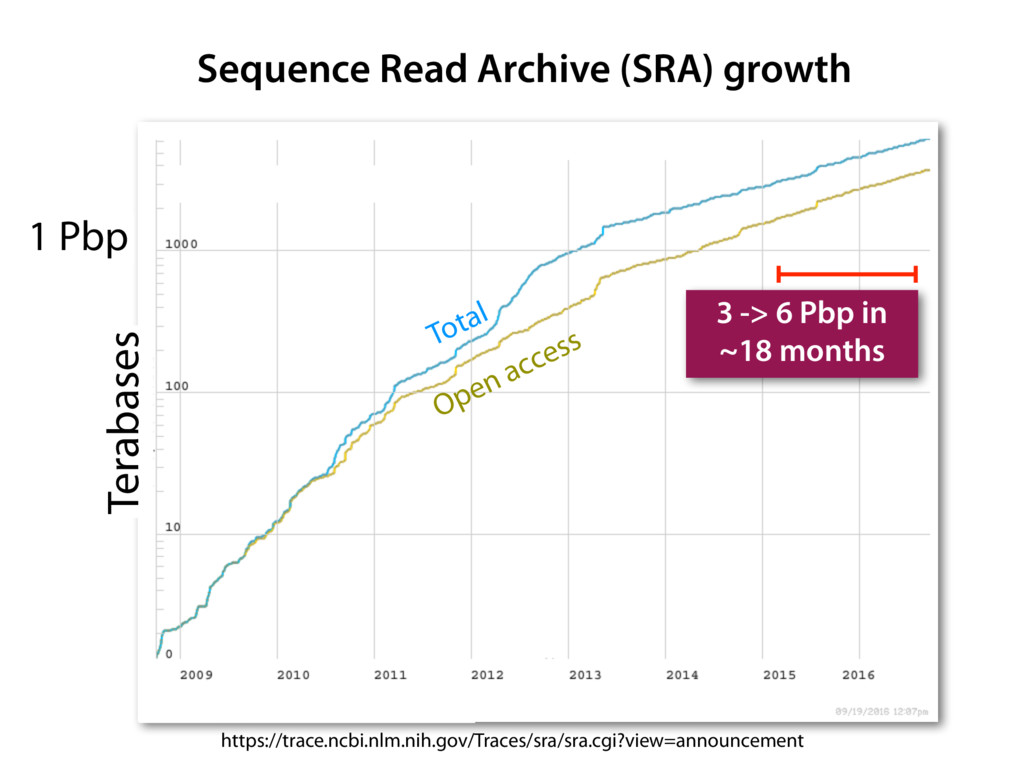
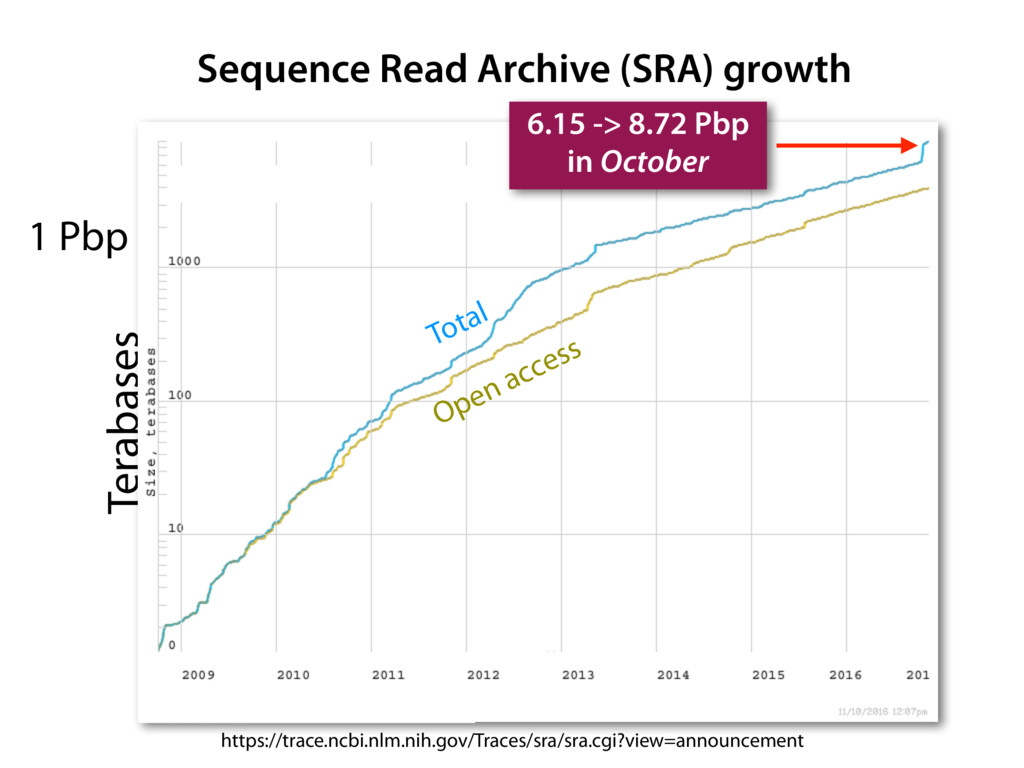
The Sequence Read Archive contains data for over 300K RNA-seq samples, including over 80K from human-derived samples. Large-scale projects like GTEx and TCGA are generating RNA-seq data on many thousands of samples. Such huge and carefully designed datasets are valuable, but unwieldy for typical biological researchers, especially when access to computational resources is limited.
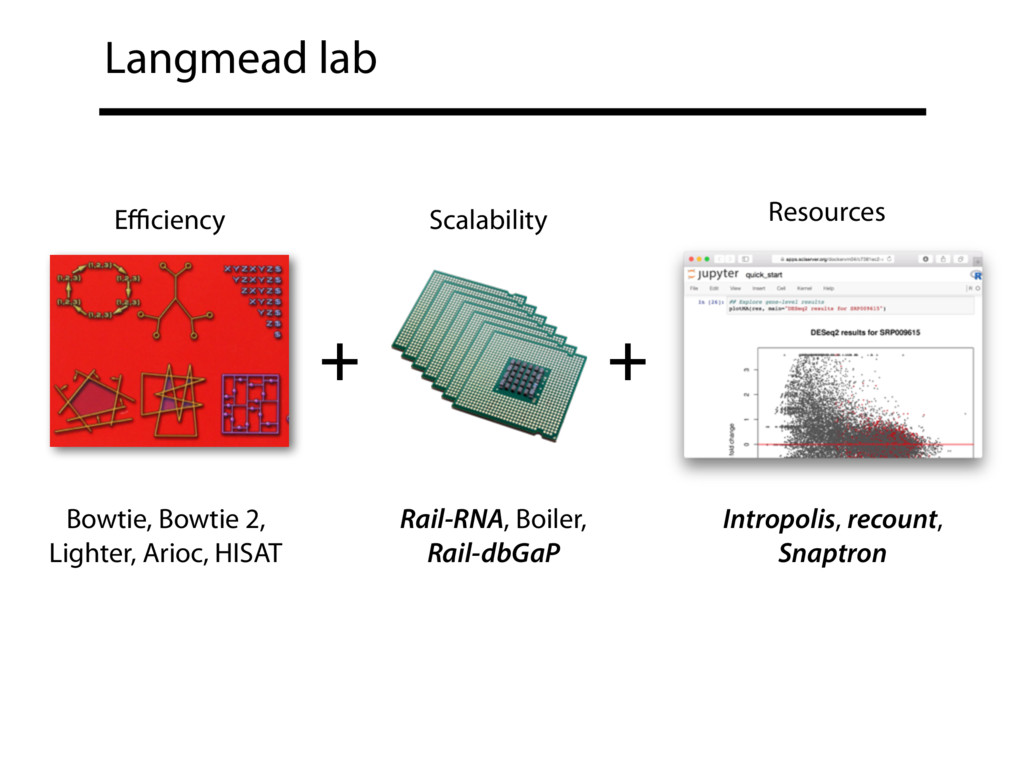
I will describe our work toward making it easy for typical biological researchers to leverage the huge amount of public RNA-seq data available today. I will highlight the Rail-RNA software (http://rail.bio), its dbGaP-protected version (http://docs.rail.bio/dbgap/), as well as the Intropolis (http://intropolis.rail.bio/) and ReCount (https://jhubiostatistics.shinyapps.io/recount/) resources. I will describe how the Rail-RNA software uses the Amazon Web Services commercial cloud to analyze many samples at once. I will also describe how we used Rail-RNA to study tens of thousands of public RNA-seq accessions, and what those studies tell us about how our knowledge of human splicing diversity has evolved over time. Finally, I will demonstrate how the Intropolis and ReCount resources can be used to pose questions about expression and differential expression across 10,000s of RNA-seq samples from the Sequence Read Archive and GTEx projects.
![Ben Langmead Assistant Professor, JHU Computer Science [email protected], langmead-lab.org, @BenLangmead](https://files.speakerdeck.com/presentations/91297d6552da4086934ca8002d9c9959/slide_0.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
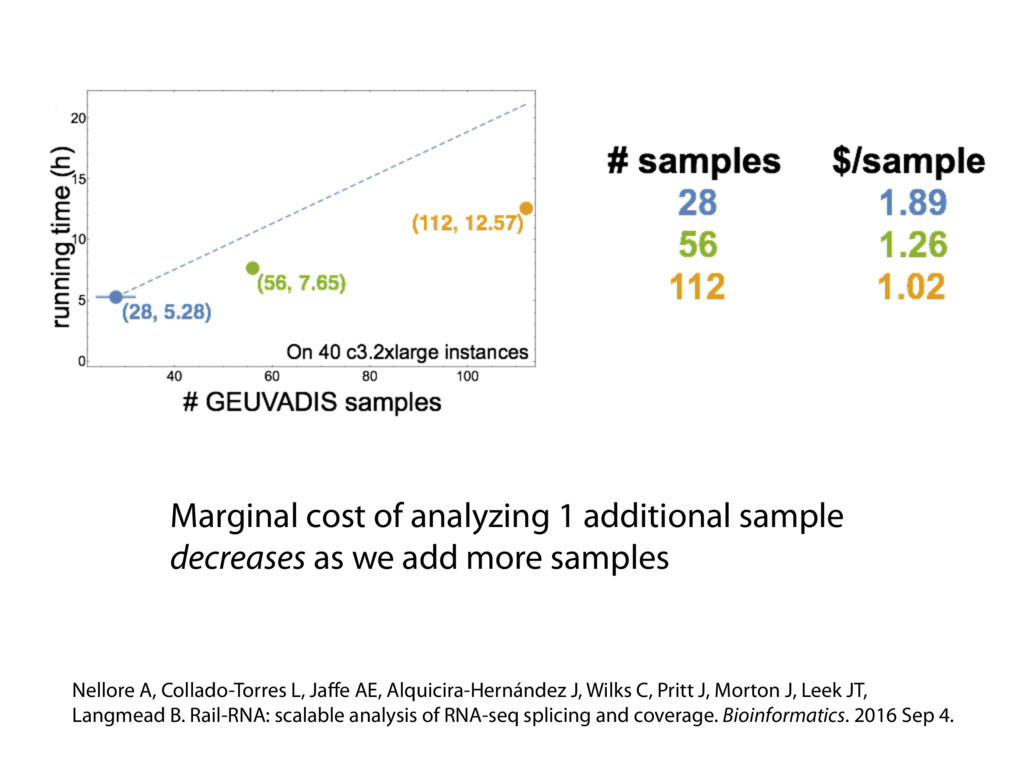{kind=link}
{kind=link}
{kind=link}
{kind=link}
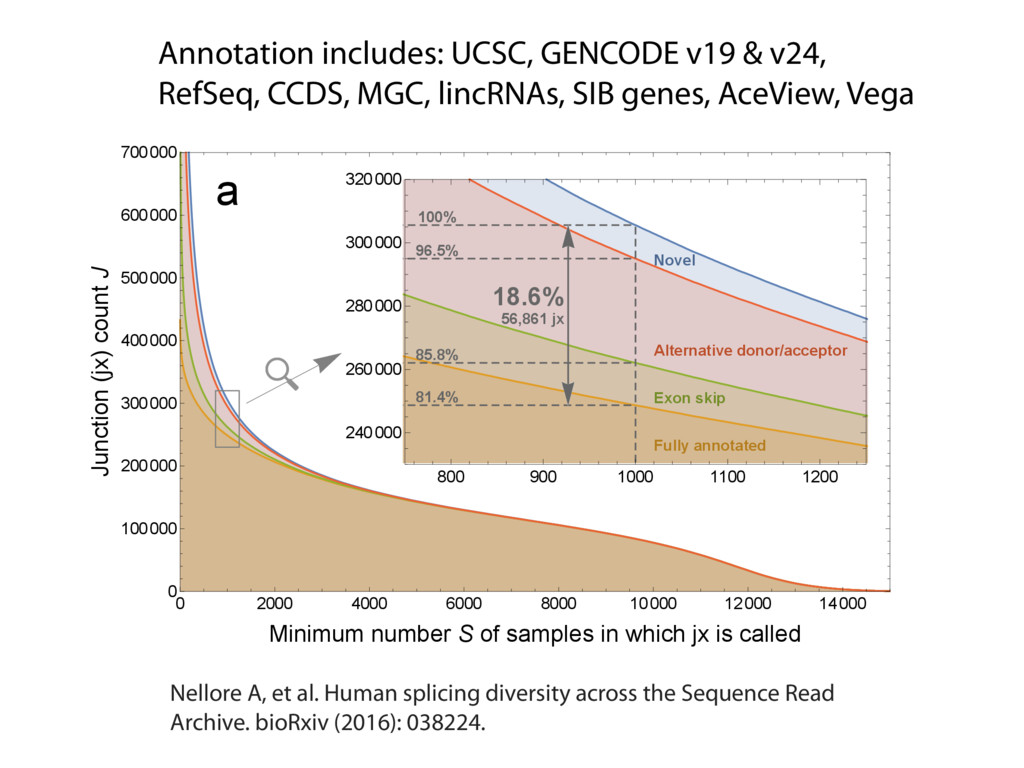{kind=link}
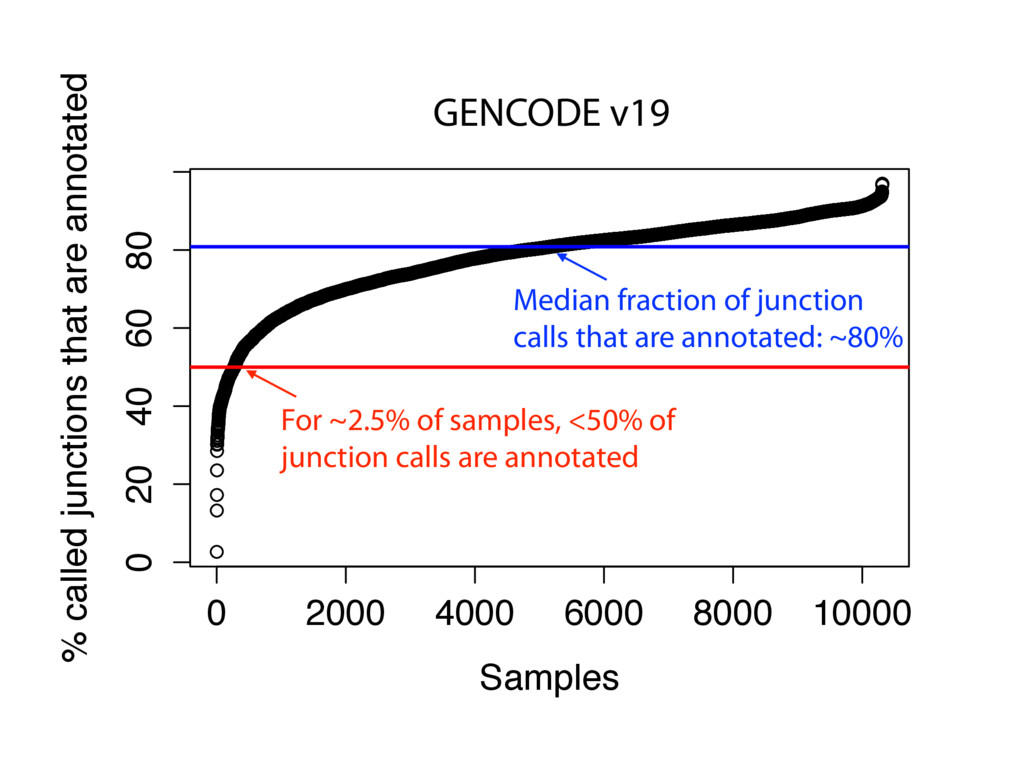{kind=link}
{kind=link}
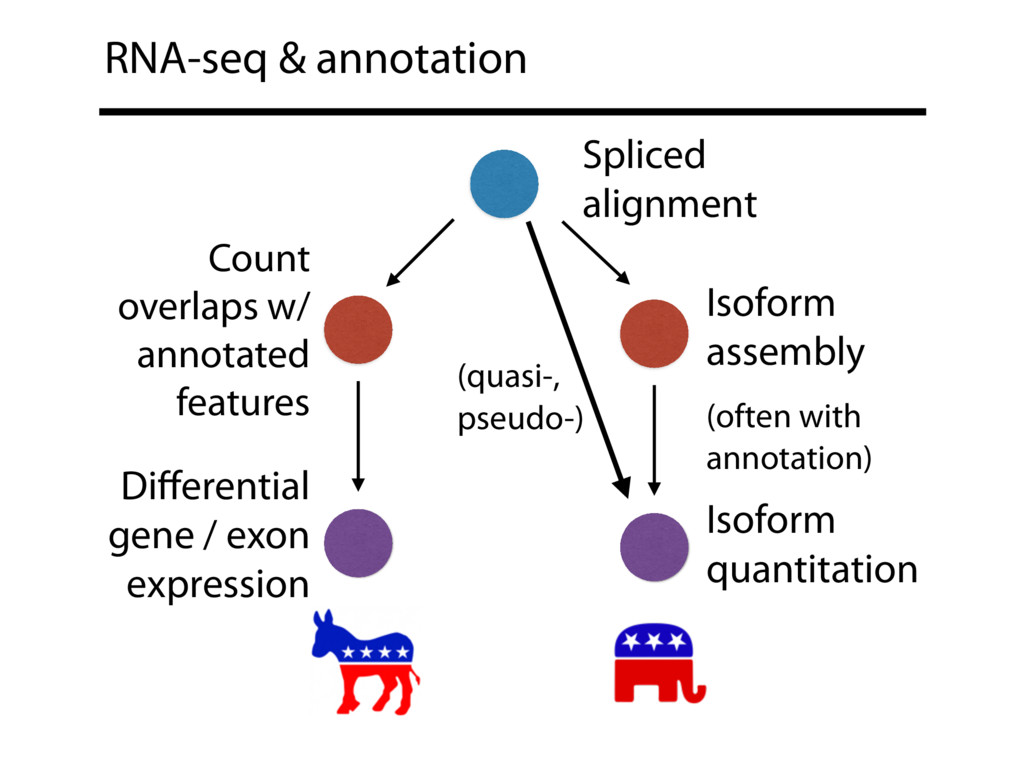{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}