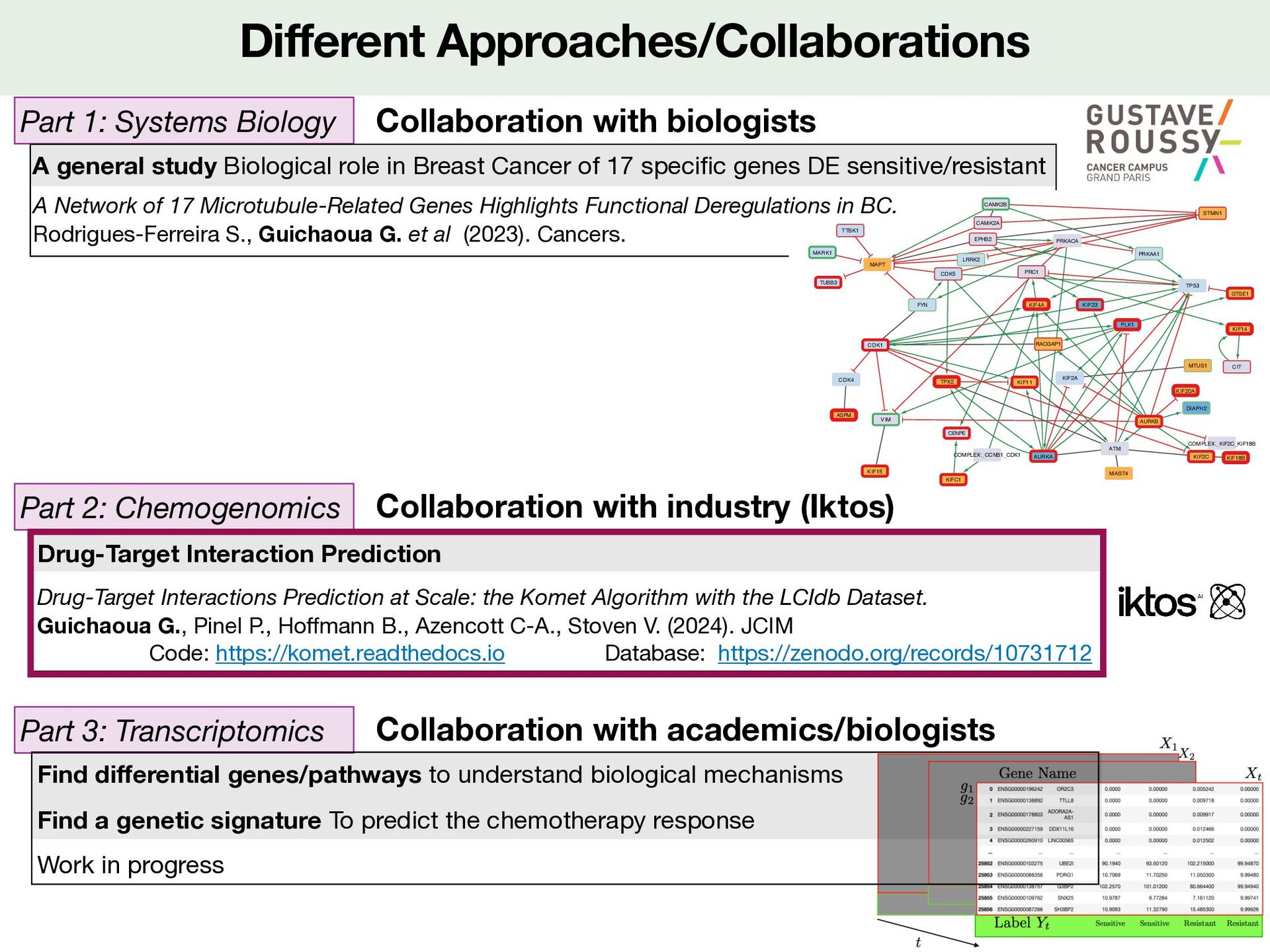
erent Approaches/Collaborations Particular ML project: Drug Target Interaction prediction General context Modelisation LCIdb: a large new training database Komet: a large scale DTI prediction method Results 2 Practice https://github.com/Guichaoua/komet/blob/main/docs/source/vignettes/komet_TP.ipynb
Master 2 Thesis 2002 2003-2022 2020-2021 2022-2025 Lycée E. Mounier (92) MVA (Mathematics, Vision and Apprentissage), ENS Paris Saclay Directed by Véronique Stoven, Chloé Azencott (CBIO - Mines Paris - Curie) Clara NAHMIAS (Institut Gustave Roussy) Machine Learning and System Biologies Approaches to fi nd new therapeutic strategies against Triple Negative Breast Cancer de fi cient in the ATIP3 protein 3
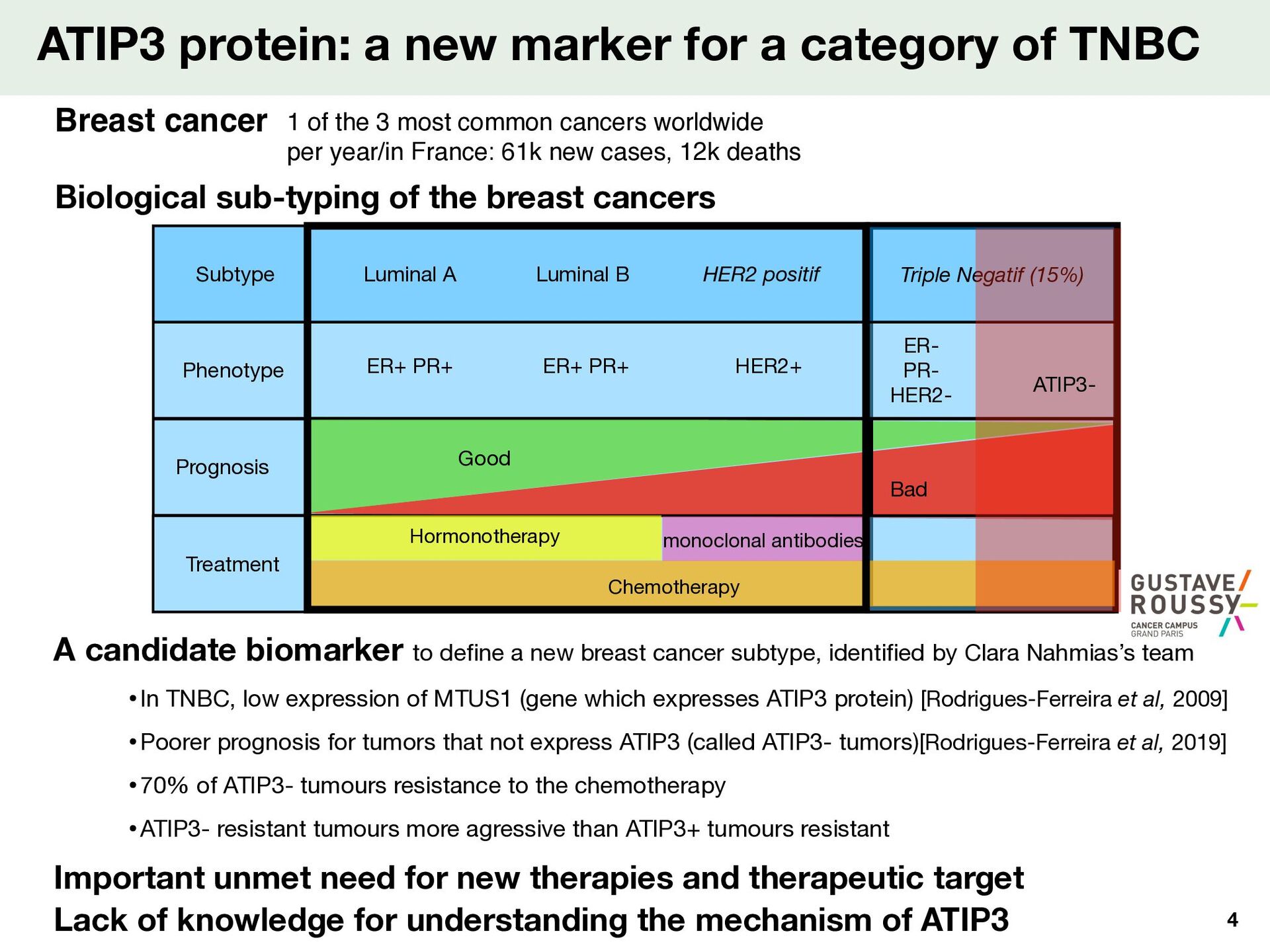
Luminal B Triple Negatif (15%) Phenotype Prognosis Treatment ER+ PR+ ER+ PR+ HER2+ ER- PR- HER2- Good ATIP3 protein: a new marker for a category of TNBC Biological sub-typing of the breast cancers Breast cancer A candidate biomarker to de fi ne a new breast cancer subtype, identi fi ed by Clara Nahmias’s team •In TNBC, low expression of MTUS1 (gene which expresses ATIP3 protein) [Rodrigues-Ferreira et al, 2009] •Poorer prognosis for tumors that not express ATIP3 (called ATIP3- tumors)[Rodrigues-Ferreira et al, 2019] •70% of ATIP3- tumours resistance to the chemotherapy •ATIP3- resistant tumours more agressive than ATIP3+ tumours resistant Important unmet need for new therapies and therapeutic target Lack of knowledge for understanding the mechanism of ATIP3 ATIP3- 4 1 of the 3 most common cancers worldwide per year/in France: 61k new cases, 12k deaths
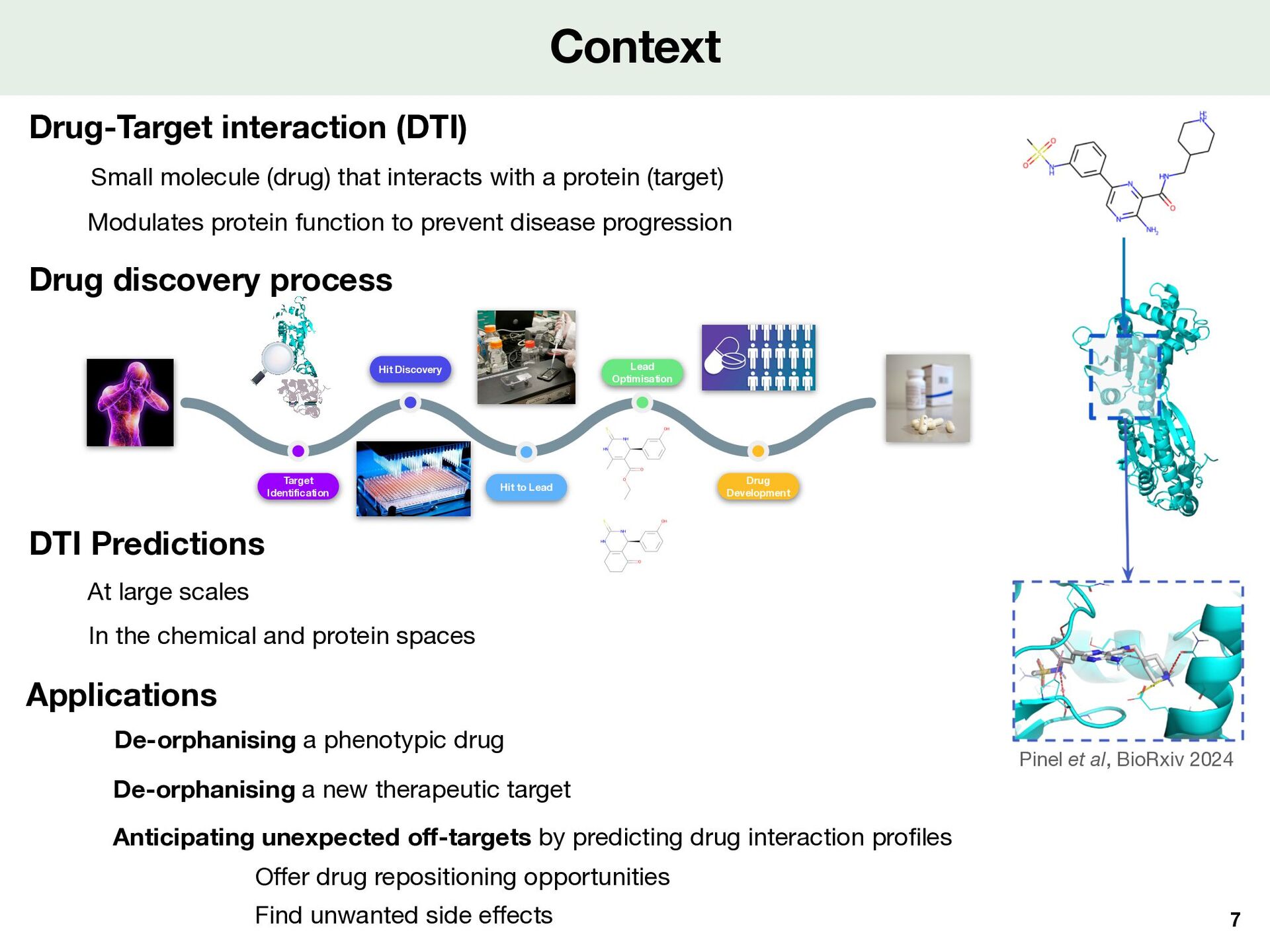
Small molecule (drug) that interacts with a protein (target) Modulates protein function to prevent disease progression Applications De-orphanising a phenotypic drug Anticipating unexpected o ff -targets by predicting drug interaction pro fi les Find unwanted side e ff ects O ff er drug repositioning opportunities De-orphanising a new therapeutic target At large scales In the chemical and protein spaces DTI Predictions Drug discovery process Hit Discovery Hit to Lead Lead Optimisation Drug Development Target Identification
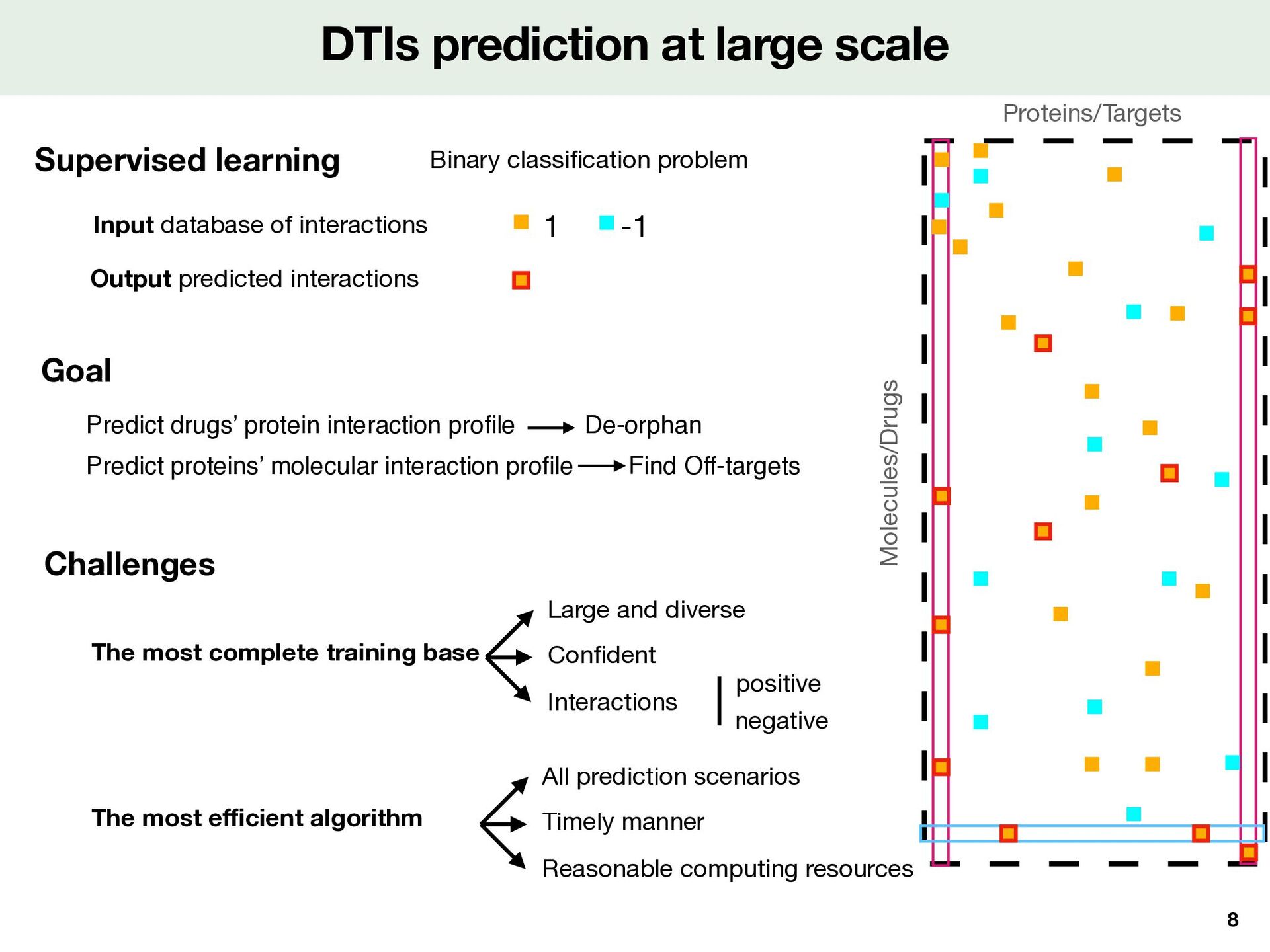
cation problem 1 -1 DTIs prediction at large scale 8 Proteins/Targets Molecules/Drugs Goal Predict drugs’ protein interaction pro fi le De-orphan Output predicted interactions Reasonable computing resources Challenges The most complete training base The most e ff i cient algorithm Large and diverse Con fi dent Interactions negative positive All prediction scenarios Timely manner Predict proteins’ molecular interaction pro fi le Find Off-targets
Construction Coverage of the protein and molecule spaces Komet: a Large-scale DTI prediction method Features of proteins and molecules Mixing for pair’s features DTI classi fi cation Results Parameters set-up of the model Impact of the mixing for pair’s features Comparison with ML algorithms
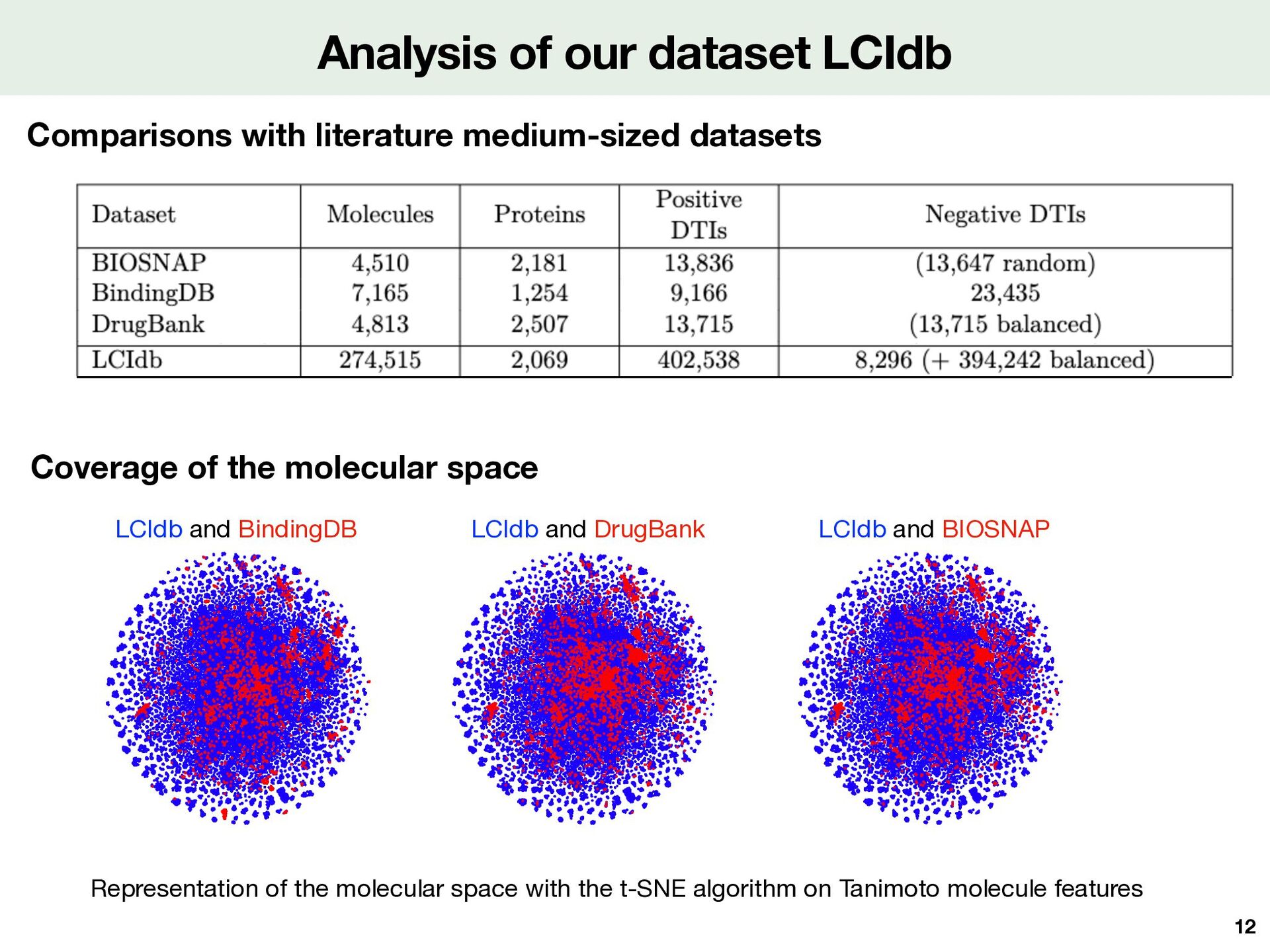
proteins 4.813 molecules 13.716 interactions + + well curated + FDA-approved drugs - only interactions + - medium-sized Protein Molecule 10 Drugbank v1.5.1 [Wishart et al, 2018] BIOSNAP [Zitnik et al, 2018] Back to bioactivity databases + Large-sized + Experimentally measures, including thermodynamic values - May have di ff erent bioactivity measures for a DTI - All molecules and proteins Kd , Ki , IC50 BindingDB [Tiqing et al, 2007], PubChem [Kim et al, 2019], ChEMBL [Mendez et al, 2019]
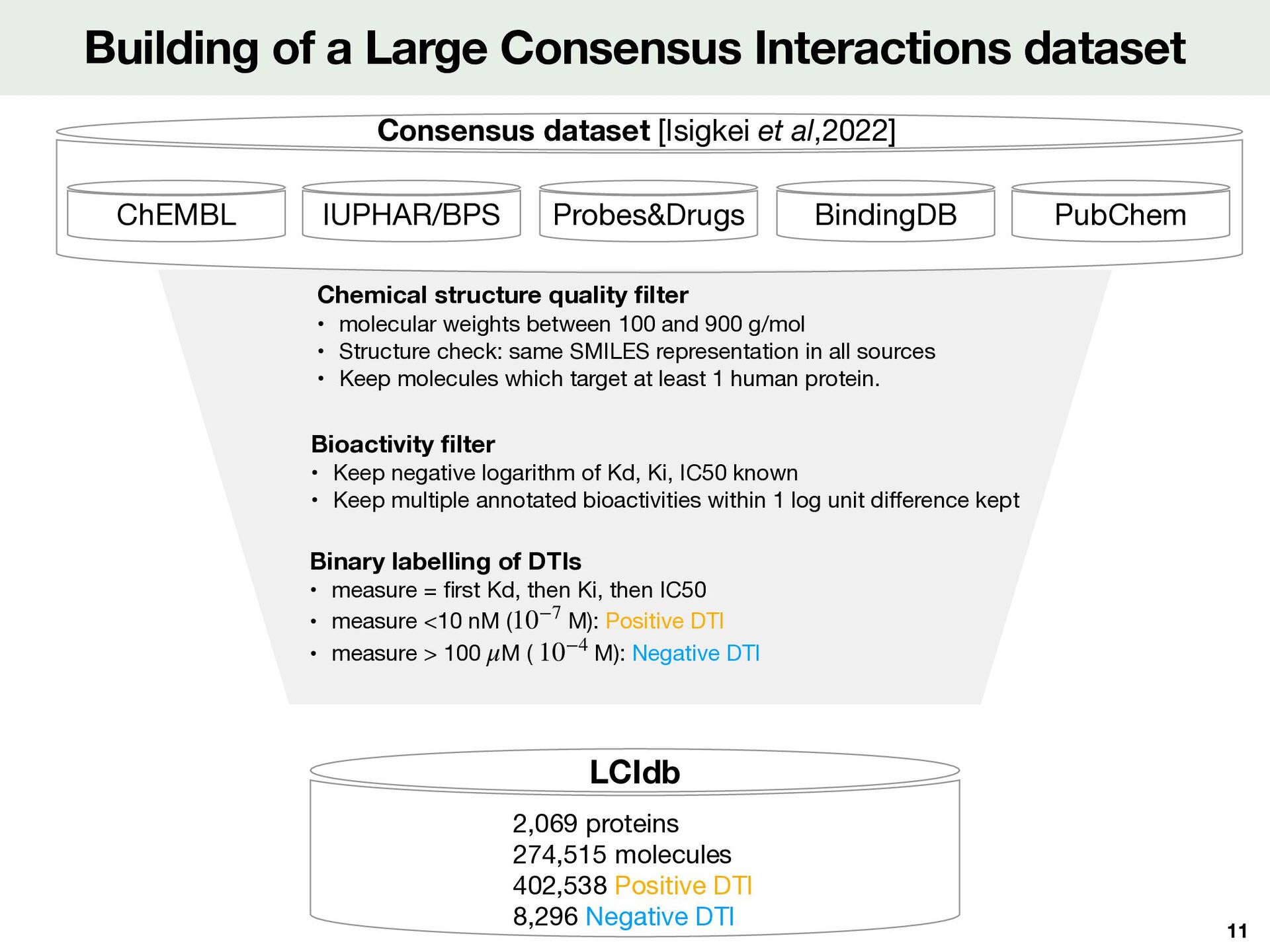
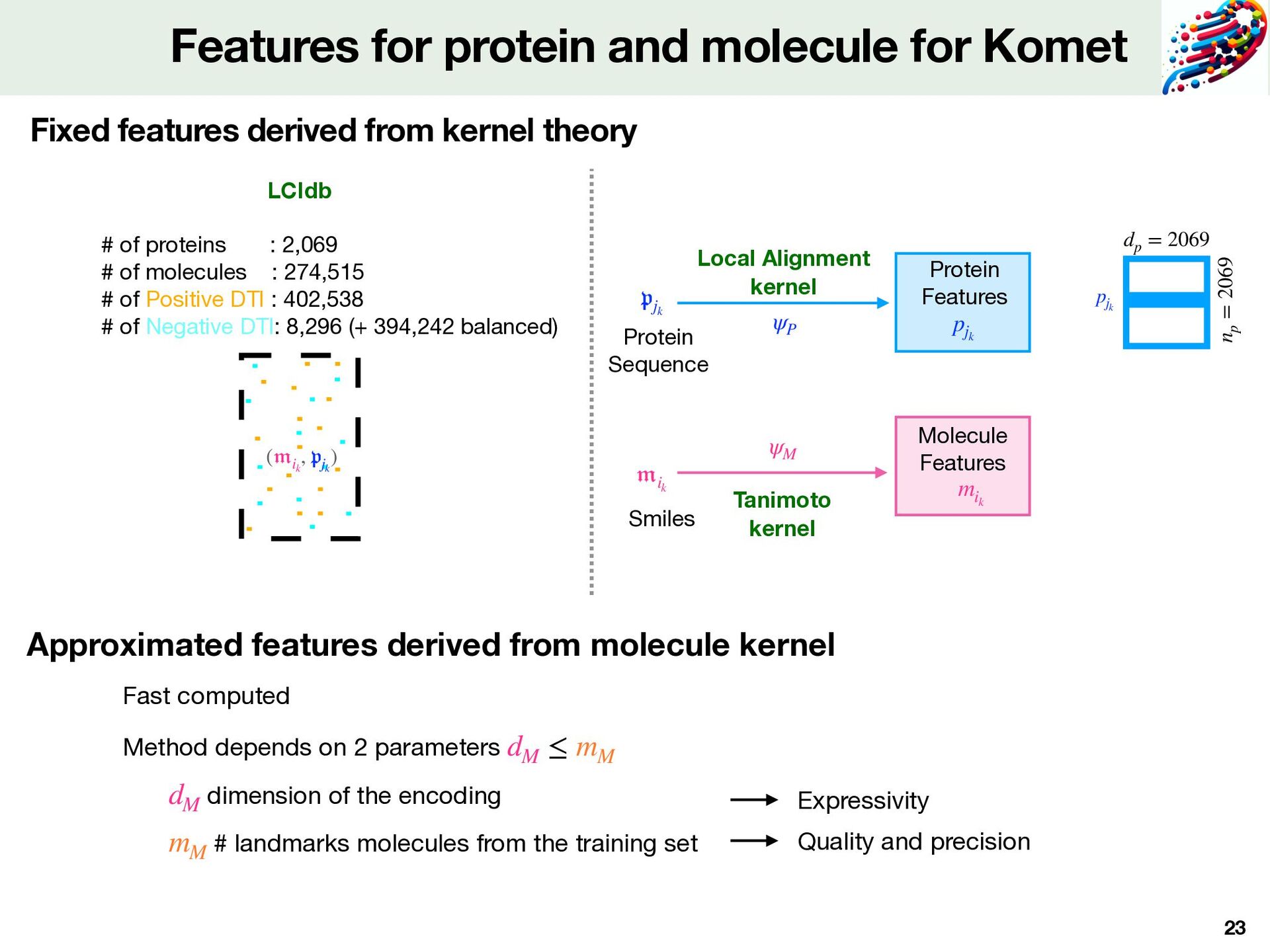
Probes&Drugs IUPHAR/BPS ChEMBL Consensus dataset [Isigkei et al,2022] Chemical structure quality fi lter • molecular weights between 100 and 900 g/mol • Structure check: same SMILES representation in all sources • Keep molecules which target at least 1 human protein. Bioactivity fi lter • Keep negative logarithm of Kd, Ki, IC50 known • Keep multiple annotated bioactivities within 1 log unit di ff erence kept Binary labelling of DTIs • measure = fi rst Kd, then Ki, then IC50 • measure <10 nM ( M): Positive DTI • measure > 100 M ( M): Negative DTI 10−7 μ 10−4 LCIdb 2,069 proteins 274,515 molecules 402,538 Positive DTI 8,296 Negative DTI
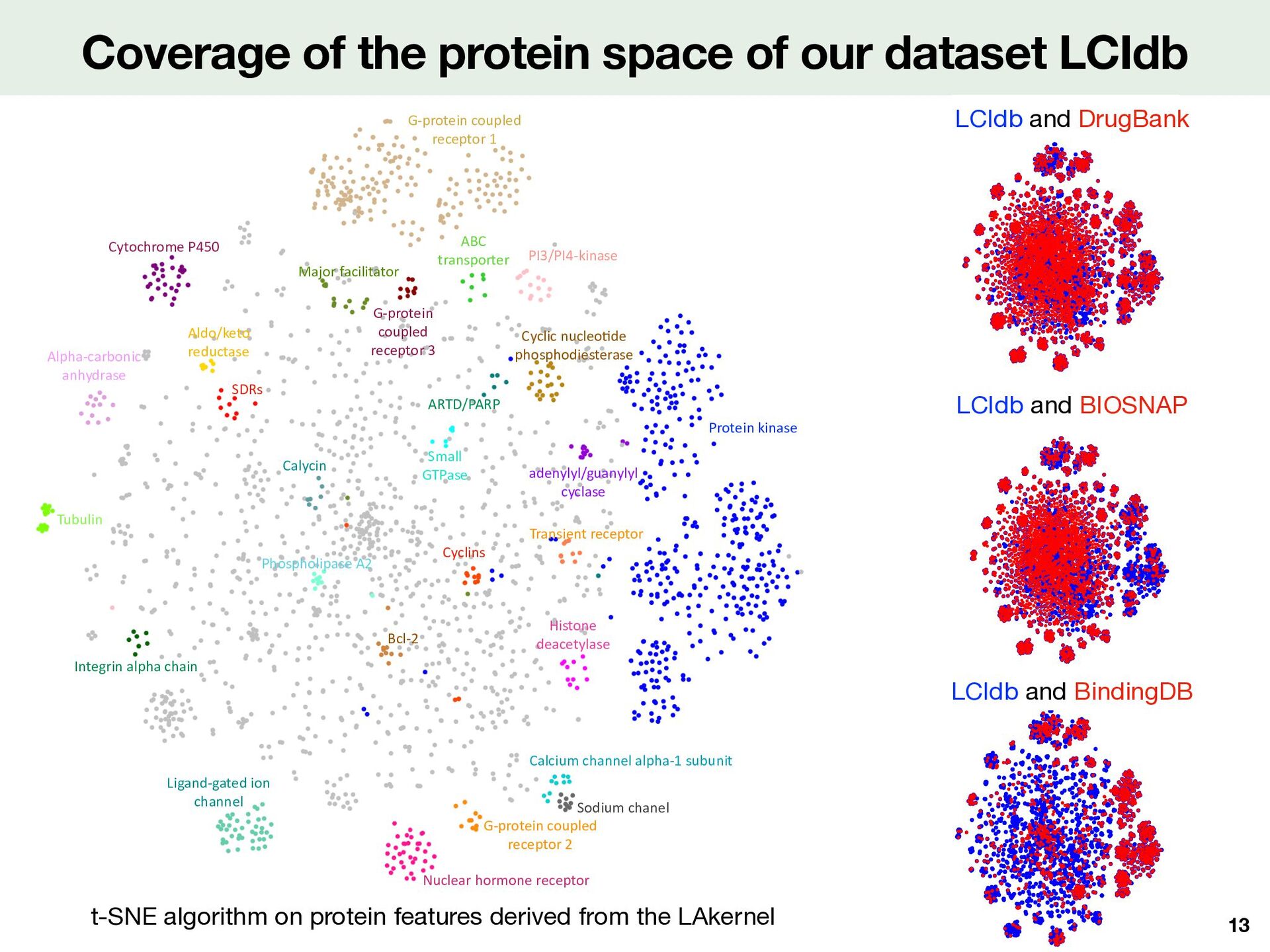
with the t-SNE algorithm on Tanimoto molecule features Comparisons with literature medium-sized datasets Coverage of the molecular space 12 LCIdb and BindingDB LCIdb and DrugBank LCIdb and BIOSNAP
molecules Mixing for pair’s features DTI classi fi cation Plan 14 LCIdb: a large new training database Motivation and Construction Coverage of the protein and molecule spaces Results Parameters set-up of the model Impact of the mixing for pair’s features Comparison of ML algorithms
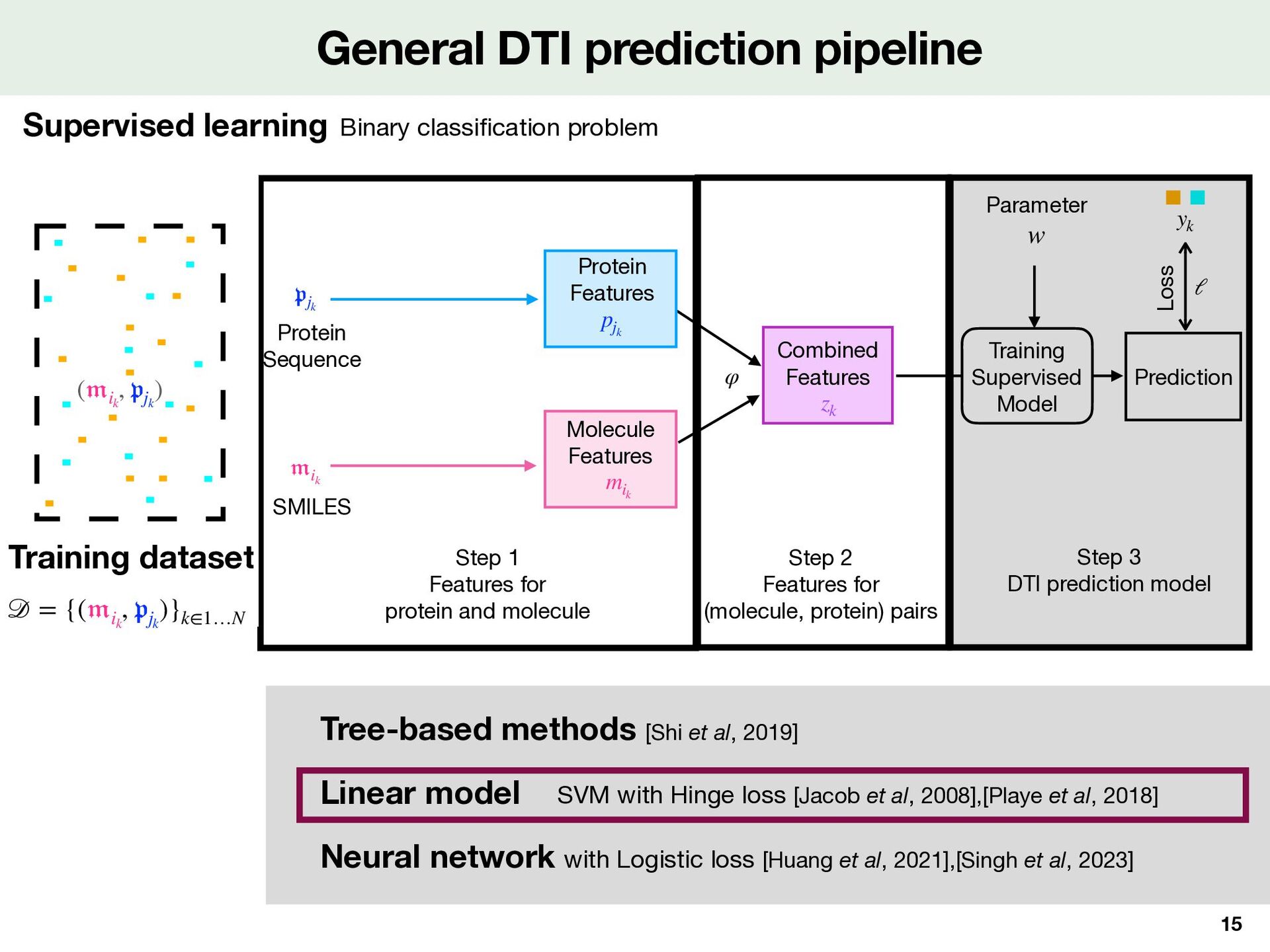
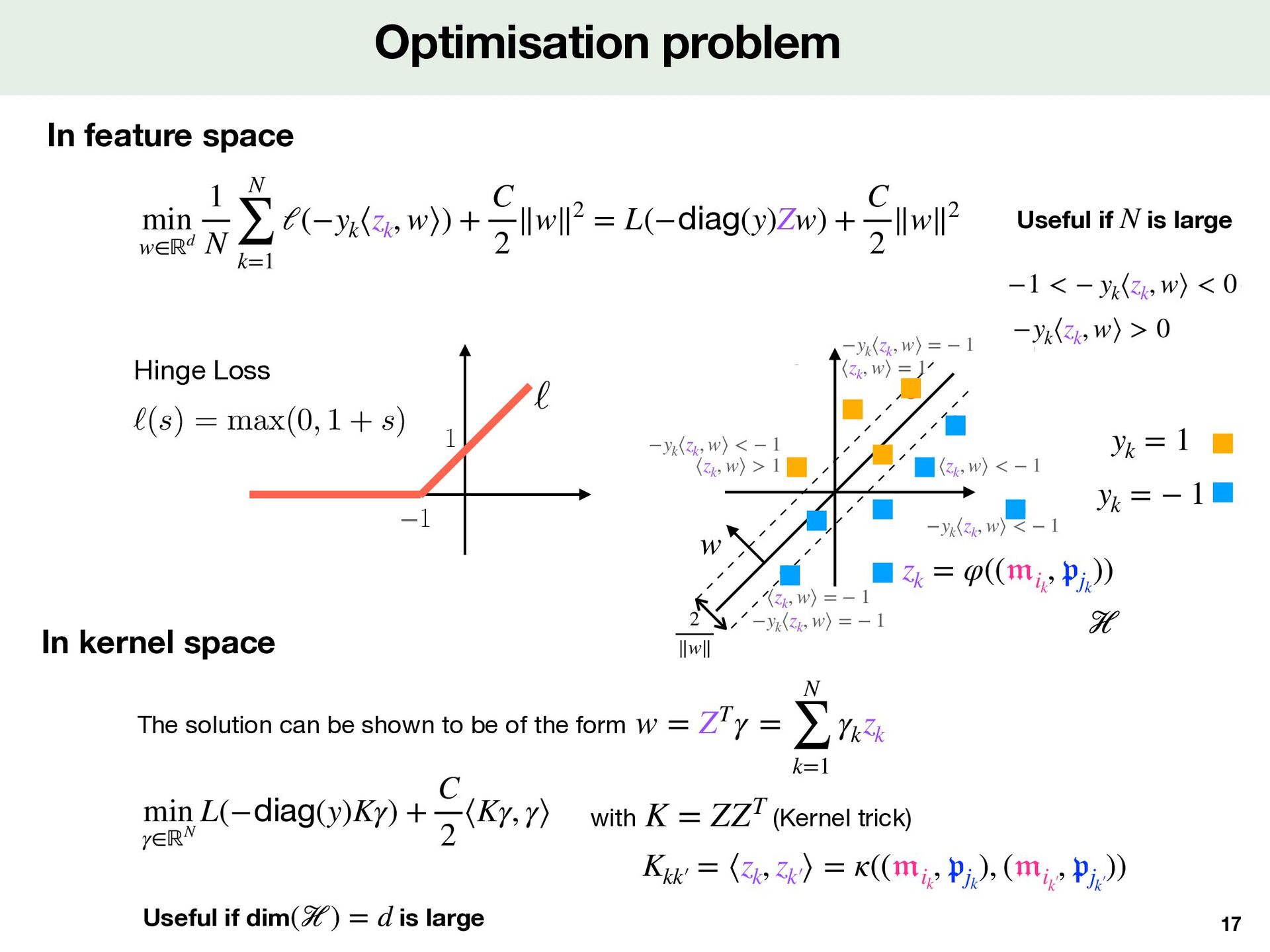
cation problem Tree-based methods [Shi et al, 2019] Neural network with Logistic loss [Huang et al, 2021],[Singh et al, 2023] SVM with Hinge loss [Jacob et al, 2008],[Playe et al, 2018] Linear model Step 1 Features for protein and molecule Step 2 Features for (molecule, protein) pairs Step 3 DTI prediction model Nystrom approximation Dimension reduction Kronecker kernel LCIdb ( ik , jk ) SMILES Protein Sequence z k Combined Features Prediction φ Loss ℓ Training Supervised Model Parameter w ik ψ M ψ P y k Local Alignment kernel Tanimoto kernel jk m ik Molecule Features Protein Features p jk # of proteins : 2,060 # of molecules : 271,180 # of Positive DTI : 396,798 # of Negative DTI: 7,965 (+ 388,833 balanced) SVM z k = m ik p⊤ jk m ik p jk z k Training dataset Training dataset 𝒟 = {( 𝔪 ik , 𝔭 jk )}k∈1…N Step 1 Features for protein and molecule Step 2 Features for (molecule, protein) pairs Step 3 DTI prediction model ( ik , jk ) SMILES Protein Sequence z k Combined Features Prediction φ Loss ℓ Training Supervised Model Parameter w ik ψ M ψ P y k Local Alignment kernel Tanimoto kernel jk m ik Molecule Features Protein Features p jk # of proteins : 2,060 # of molecules : 271,180 # of Positive DTI : 396,798 # of Negative DTI: 7,965 (+ 388,833 balanced) z k = m ik p⊤ jk m ik p jk z k Training dataset
Kernel point of view de fi nes a kernel , a similarity between two pairs and φ κ ( 𝔪 , 𝔭 ) ( 𝔪 ′  , 𝔭 ′  ) κ(( 𝔪 , 𝔭 ), ( 𝔪 ′  , 𝔭 ′  )) := ⟨φ(( 𝔪 , 𝔭 )), φ(( 𝔪 ′  , 𝔭 ′  ))⟩ℋ yk = 1 yk = − 1 zk = φ(( 𝔪 ik , 𝔭 jk )) Step 1 Features for protein and molecule Step 2 Features for (molecule, protein) pairs Step 3 DTI prediction model Dimension reduction kernel ( ik , jk ) SMILES Protein Sequence z k Combined Features Prediction φ Loss ℓ Training Supervised Model Parameter w ik ψ M ψ P y k Local Alignment kernel Tanimoto kernel jk m ik Molecule Features Protein Features p jk # of proteins : 2,060 # of molecules : 271,180 # of Positive DTI : 396,798 # of Negative DTI: 7,965 (+ 388,833 balanced) z k = m ik p⊤ jk m ik p jk z k Training dataset 𝒟 = {( 𝔪 ik , 𝔭 jk )}k∈1…N φ ℋ w f( 𝔪 , 𝔭 ) = ⟨φ(( 𝔪 , 𝔭 )), w⟩ℋ f > 0 f < 0 Linear classi fi er in features space Note that we can also start by and de fi ne a κ φ Useful if dim( ) is large, even in fi nite ℋ
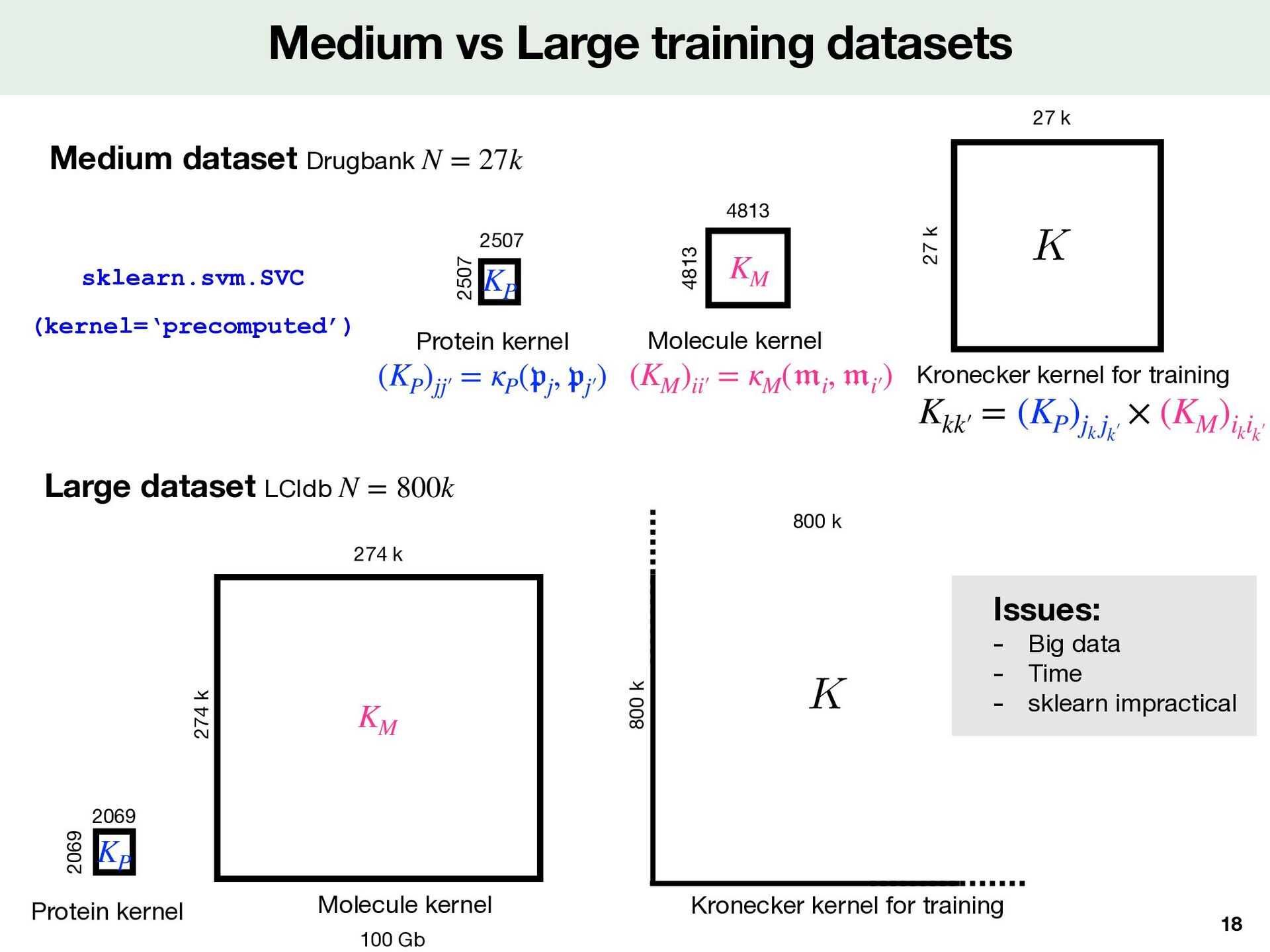
27k sklearn.svm.SVC (kernel=‘precomputed’) 18 27 k 27 k 2507 2507 Protein kernel 4813 4813 Molecule kernel Kronecker kernel for training <latexit sha1_base64="JhUk4wXzwRCmCXl9jAE1rQYQvmU=">AAACxnicjVHLSsNAFD2NrxpfVZdugkVwVRLxtSy6KbipaB9QiyTptA7Ni8lEKUXwB9zqp4l/oH/hnTEFtYhOSHLm3HvOzL3XSwKeStt+LRgzs3PzC8VFc2l5ZXWttL7RTONM+Kzhx0Es2p6bsoBHrCG5DFg7EcwNvYC1vOGpirdumUh5HF3KUcK6oTuIeJ/7riTq4sw0r0tlu2LrZU0DJwdl5Ksel15whR5i+MgQgiGCJBzARUpPBw5sJMR1MSZOEOI6znAPk7QZZTHKcIkd0ndAu07ORrRXnqlW+3RKQK8gpYUd0sSUJwir0ywdz7SzYn/zHmtPdbcR/b3cKyRW4obYv3STzP/qVC0SfRzrGjjVlGhGVefnLpnuirq59aUqSQ4JcQr3KC4I+1o56bOlNamuXfXW1fE3nalYtffz3Azv6pY0YOfnOKdBc6/iHFYOzvfL1ZN81EVsYRu7NM8jVFFDHQ3yHuART3g2akZkZMbdZ6pRyDWb+LaMhw9/Ho+C</latexit> K Kkk′  = (KP )jk jk′  × (KM )ik ik′  (KP )jj′  = κP ( 𝔭 j , 𝔭 j′  ) KP (KM )ii′  = κM ( 𝔪 i , 𝔪 i′  ) KM Large dataset LCIdb N = 800k 274 k 274 k 100 Gb Molecule kernel Issues: - Big data - Time - sklearn impractical 800 k 800 k Kronecker kernel for training <latexit sha1_base64="JhUk4wXzwRCmCXl9jAE1rQYQvmU=">AAACxnicjVHLSsNAFD2NrxpfVZdugkVwVRLxtSy6KbipaB9QiyTptA7Ni8lEKUXwB9zqp4l/oH/hnTEFtYhOSHLm3HvOzL3XSwKeStt+LRgzs3PzC8VFc2l5ZXWttL7RTONM+Kzhx0Es2p6bsoBHrCG5DFg7EcwNvYC1vOGpirdumUh5HF3KUcK6oTuIeJ/7riTq4sw0r0tlu2LrZU0DJwdl5Ksel15whR5i+MgQgiGCJBzARUpPBw5sJMR1MSZOEOI6znAPk7QZZTHKcIkd0ndAu07ORrRXnqlW+3RKQK8gpYUd0sSUJwir0ywdz7SzYn/zHmtPdbcR/b3cKyRW4obYv3STzP/qVC0SfRzrGjjVlGhGVefnLpnuirq59aUqSQ4JcQr3KC4I+1o56bOlNamuXfXW1fE3nalYtffz3Azv6pY0YOfnOKdBc6/iHFYOzvfL1ZN81EVsYRu7NM8jVFFDHQ3yHuART3g2akZkZMbdZ6pRyDWb+LaMhw9/Ho+C</latexit> K 2069 2069 Protein kernel KP KM
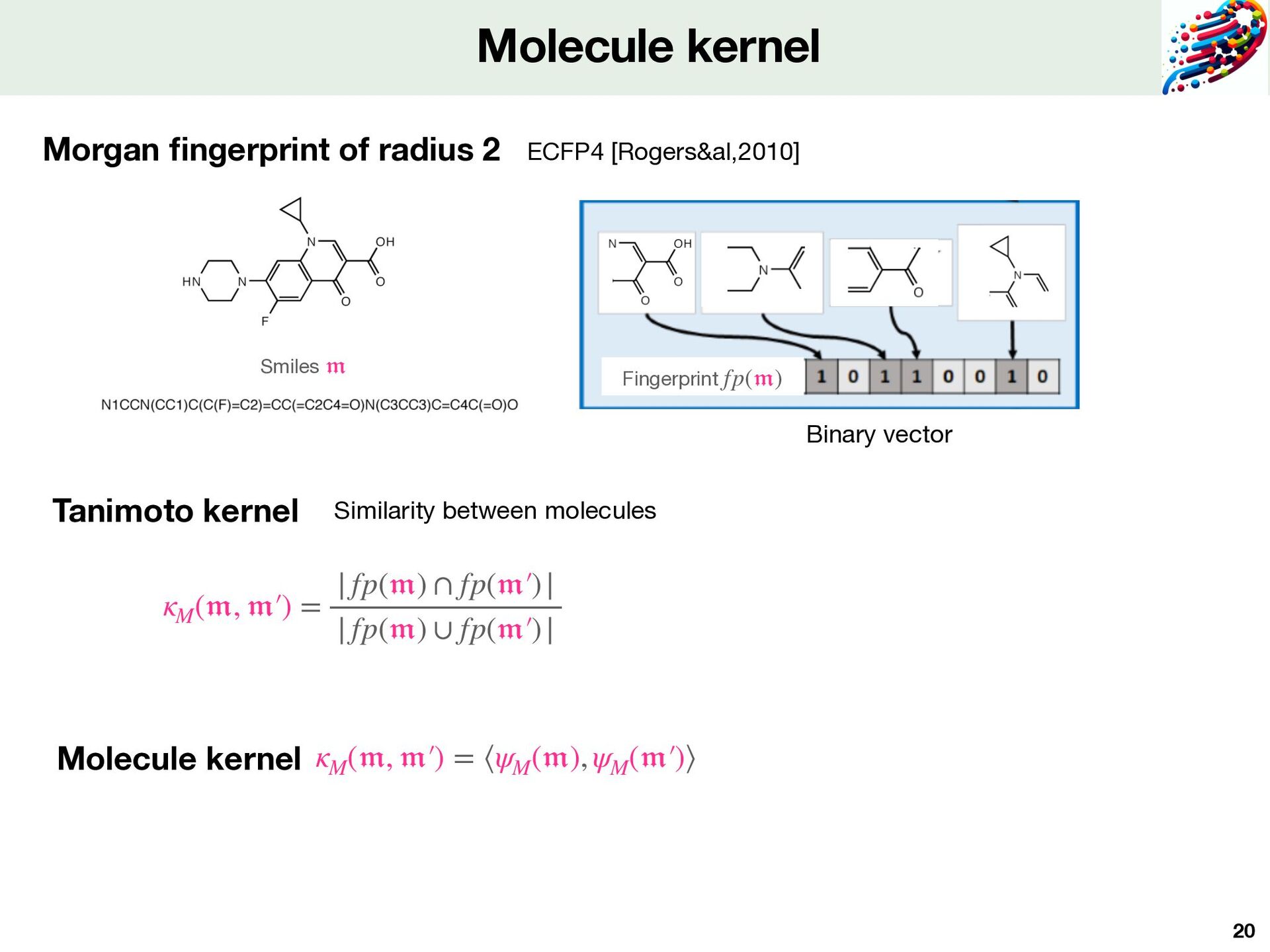
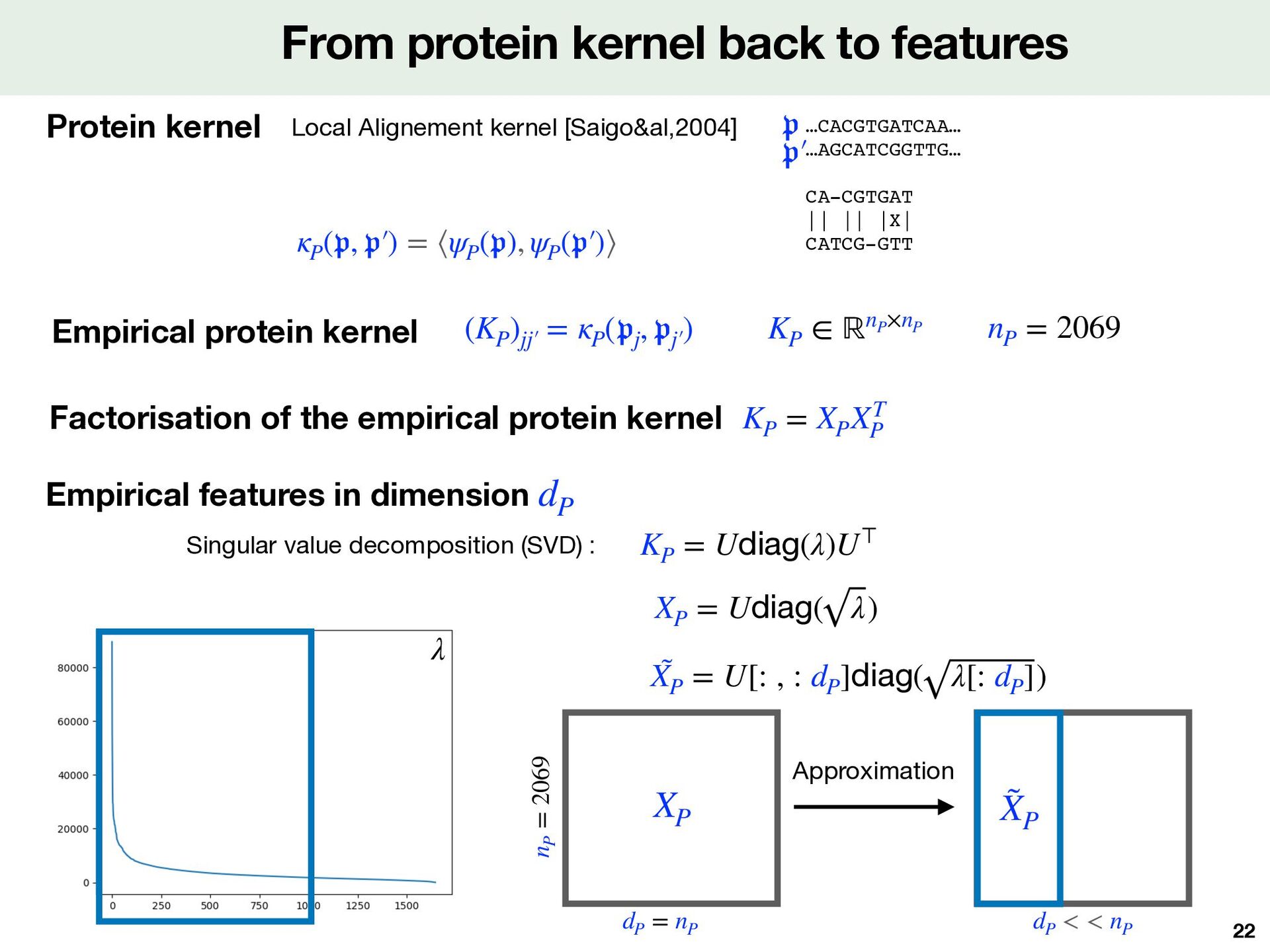
kernel theory Note that we can use learned features Features compatible with large-scale Computed on simple representations for protein and molecule 𝔪 Smiles MAGPSLACCLLGLLALTSACYIQNCPLGGKRAAPDLDVRKCLPCGPGGKG RCFGPNICCAEELGCFVGTAEALRCQEENYLPSPCQSGQKACGSGGRCAV LGLCCSPDGCHADPACDAEATFSQR 𝔭 Sequence of amino acids Morgan fi ngerprints ECFP4 [Rogers&al,2010] Tanimoto kernel ⟶ Local Alignement kernel [Saigo&al,2004] …CACGTGATCAA… …AGCATCGGTTG… 𝔭 𝔭 ′  CA-CGTGAT || || |X| CATCG-GTT
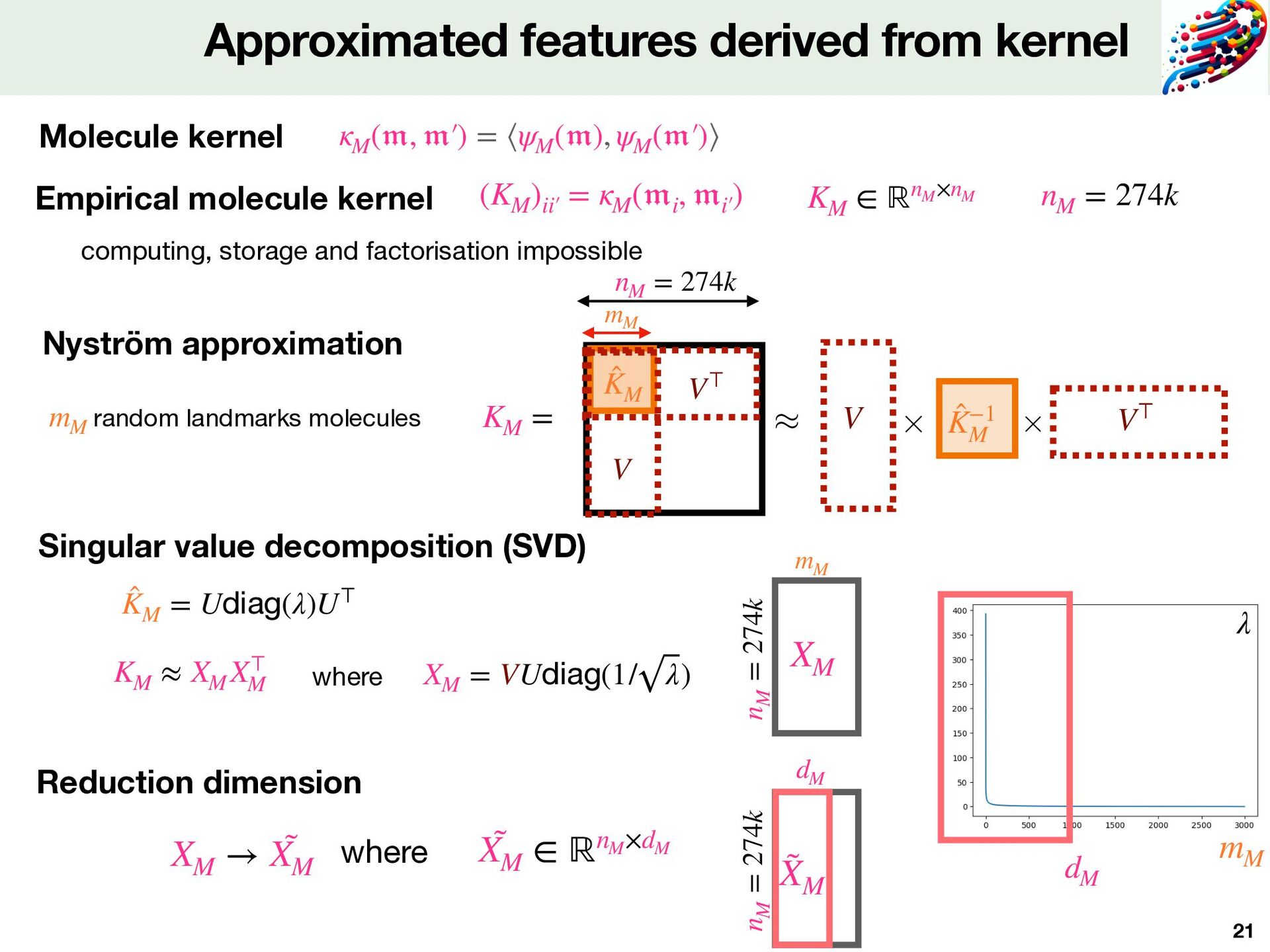
and molecule for Komet Protein Sequence 𝔪 ik ψM ψP Local Alignment kernel Tanimoto kernel 𝔭 jk mik Molecule Features Protein Features pjk Smiles ( 𝔪 ik , 𝔭 jk ) # of proteins : 2,069 # of molecules : 274,515 # of Positive DTI : 402,538 # of Negative DTI: 8,296 (+ 394,242 balanced) LCIdb Fixed features derived from kernel theory Fast computed Method depends on 2 parameters dM ≤ mM dimension of the encoding dM # landmarks molecules from the training set mM np = 2069 pjk dp = 2069 Expressivity Quality and precision
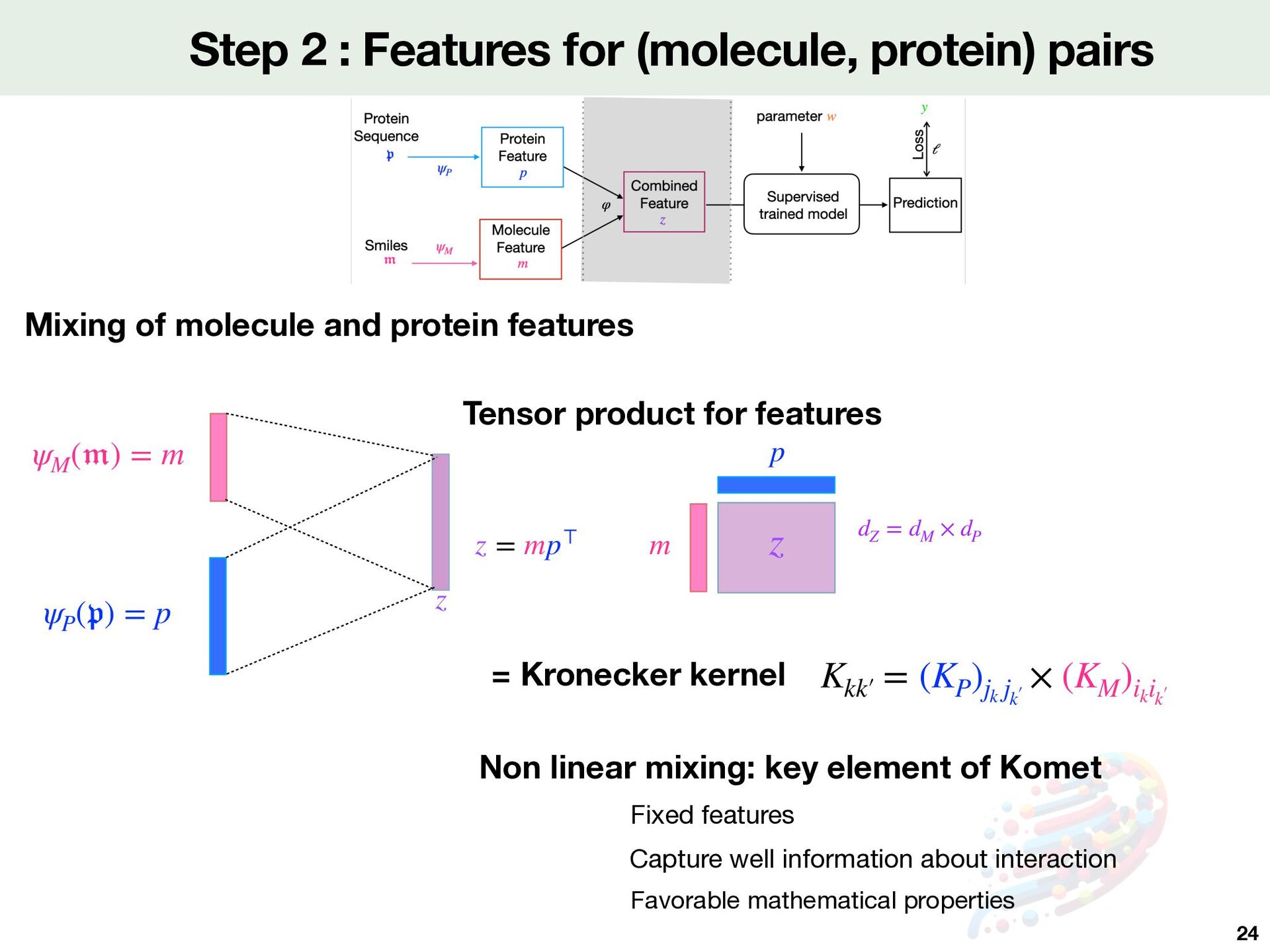
( 𝔭 ) = p z Mixing of molecule and protein features ψM ( 𝔪 ) = m Capture well information about interaction Favorable mathematical properties Non linear mixing: key element of Komet Fixed features z = mp⊤ m p z dZ = dM × dP Tensor product for features Kkk′  = (KP )jk jk′  × (KM )ik ik′  = Kronecker kernel
⟩ℝdZ = ⟨w, mik p⊤ jk ⟩ℝdP×dM = ⟨Wpjk , mik ⟩ℝdM E ffi cient computation [Airola&Pahikkala,2017] can be computed in only operations (qj )nP j=1 nP × dZ ⏟ qjk Complexity Explicit Zw Implicit computation nZ × dZ nP × dZ + nZ × dM Problem is too big for both storage and computation of Z Zw Z ∈ ℝnZ ×dZ zk nZ = 460k dZ = dM × dP GPU Code in PyTorch Full batch BFGS method to solve the optimisation problem Optimisation problem min w∈ℝ(dP×dM) nZ ∑ k=1 ℓ(⟨w, zk ⟩, yk ) + C 2 ∥w∥2 SVM https://komet.readthedocs.io
Construction Coverage of the protein and molecule spaces Komet: a Large-scale DTI prediction method Features of proteins and molecules Mixing for pair’s features DTI classi fi cation Results Parameters set-up of the model Impact of the mixing for pair’s features Comparison of ML algorithms
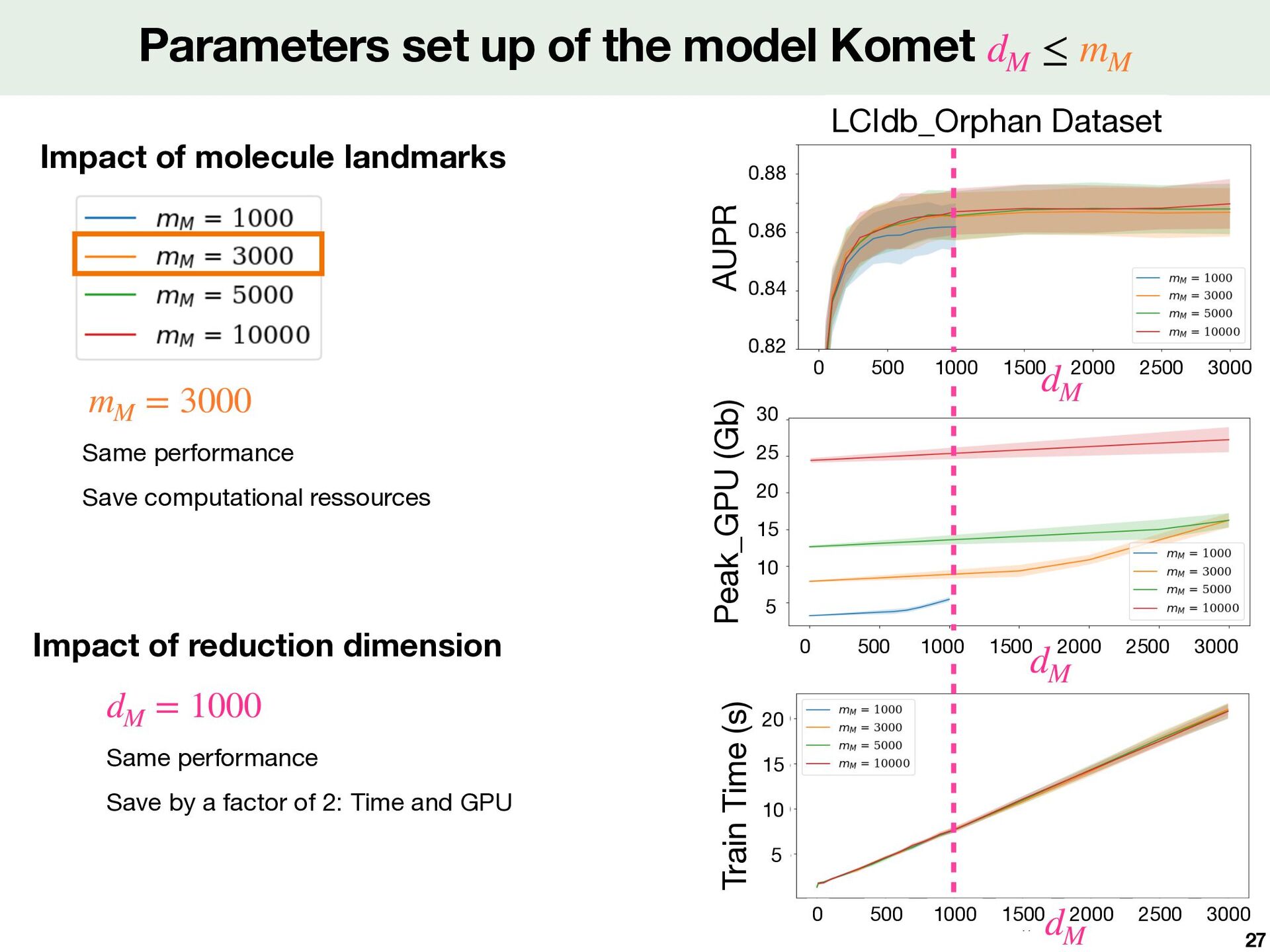
of the model Komet dM ≤ mM Peak_GPU (Gb) Impact of reduction dimension Train Time (s) Same performance Save by a factor of 2: Time and GPU dM = 1000 Same performance Save computational ressources mM = 3000 0 500 1000 1500 2000 2500 3000 dM 0.82 0.84 0.86 0.88 0 500 1000 1500 2000 2500 3000 5 15 20 25 10 30 dM 0 500 1000 1500 2000 2500 3000 dM 5 15 20 10 27
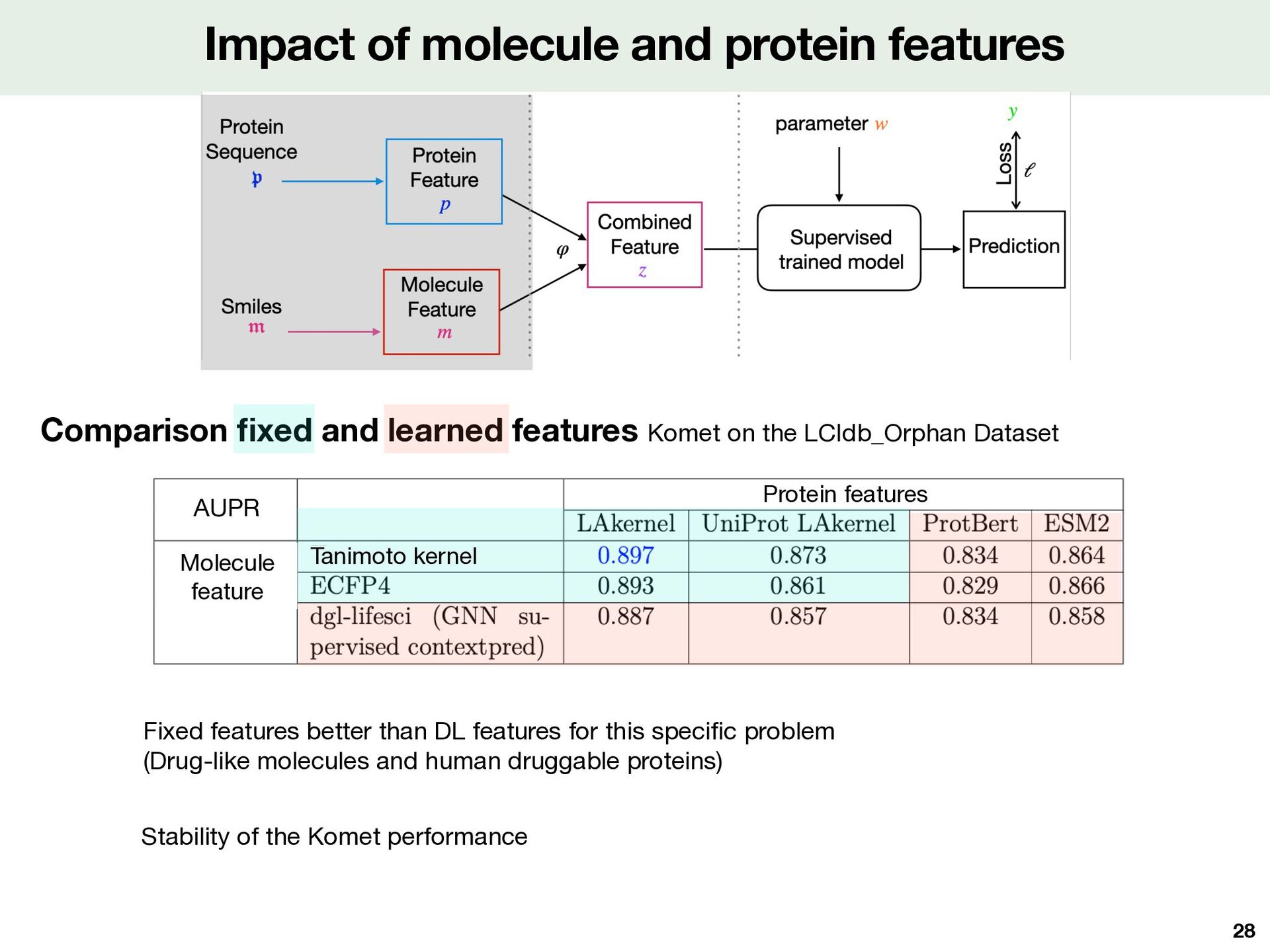
learned features Komet on the LCIdb_Orphan Dataset Fixed features better than DL features for this speci fi c problem (Drug-like molecules and human druggable proteins) Stability of the Komet performance dM = 1000 mM = 3000 Protein features Tanimoto kernel Molecule feature AUPR 28
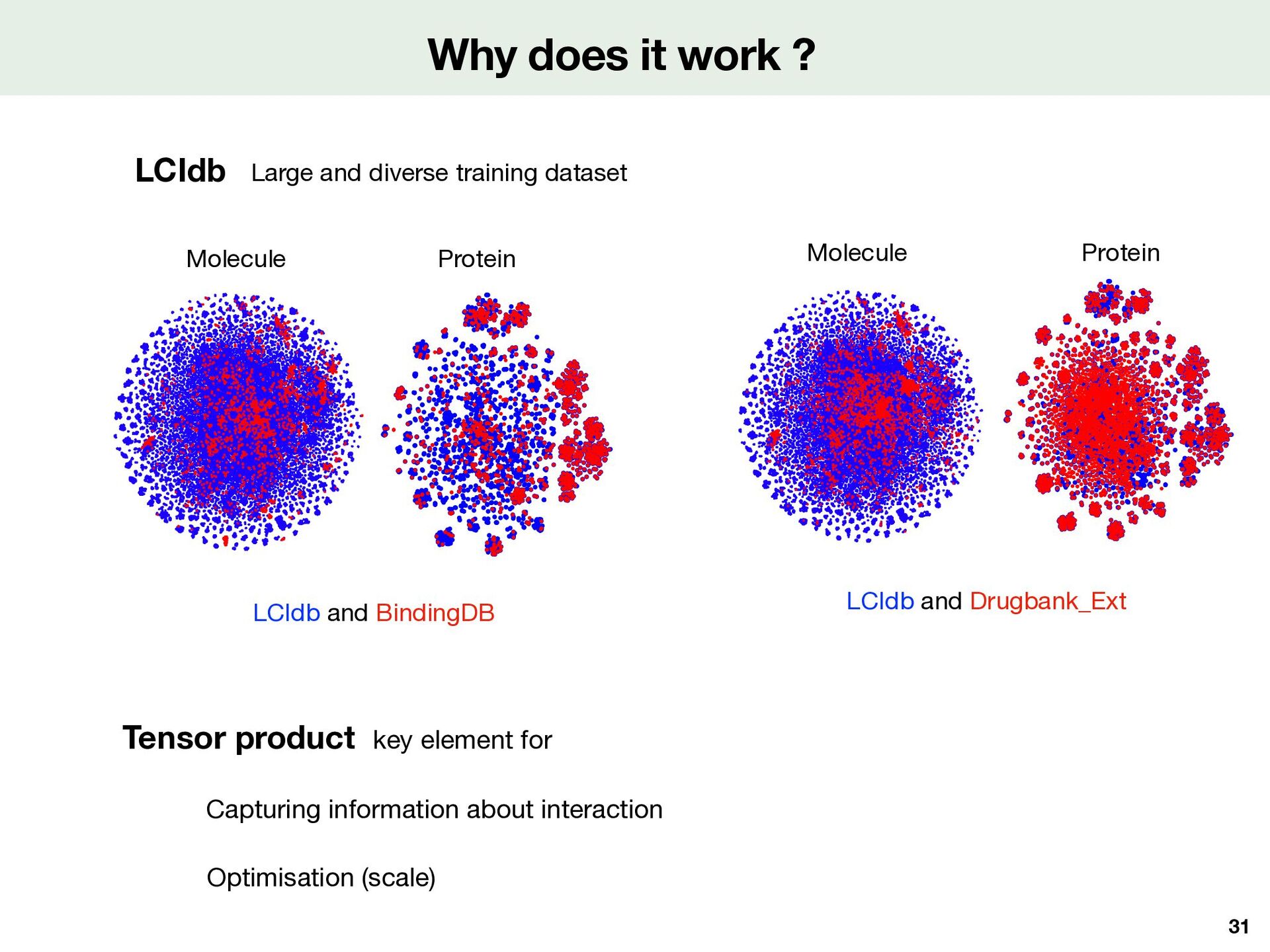
BIOSNAP BindingDB LCIdb LCIdb Drugbank Optimisation (scale) Capturing information about interaction Large and diverse training dataset LCIdb and BindingDB LCIdb and Drugbank_Ext Molecule Molecule Protein Protein 31
protein and molecule spaces Contributions: A large new molecule/protein interactions dataset https://zenodo.org/records/10731712 Komet: Fast & State of the Art https://komet.readthedocs.io Perspectives: Analysis of the target proteins predicted for the molecules found in phenotypic drug screening
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![Kernel SVM where trained by SVM [Vert&al, 2008] w 16](https://files.speakerdeck.com/presentations/349fb793a3e944a2ab87c6f2cf4fdabb/slide_15.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}