String Graph Assembly For Diploid Genomes With Long Reads
This is from the talk I gave in CSHL Genomic Informatics Meeting 2013. It demonstrates an algorithm based on string graph for assembling diploid from PacBio(R) single molecule reads larger than 10kb.
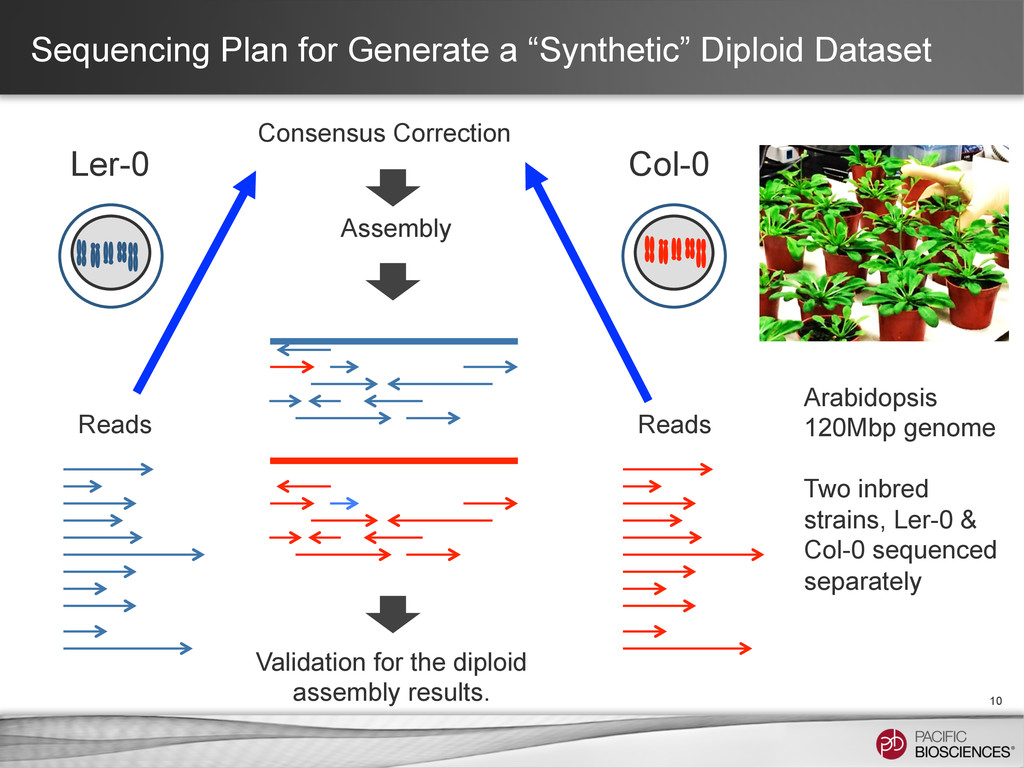
of California, Inc. All rights reserved. Jason Chin, Paul Peluso & David Rank, Pacific Biosciences Cold Spring Harbor Laboratory, Genome Informatics 2013 String Graph Assembly For Diploid Genomes With Long Reads
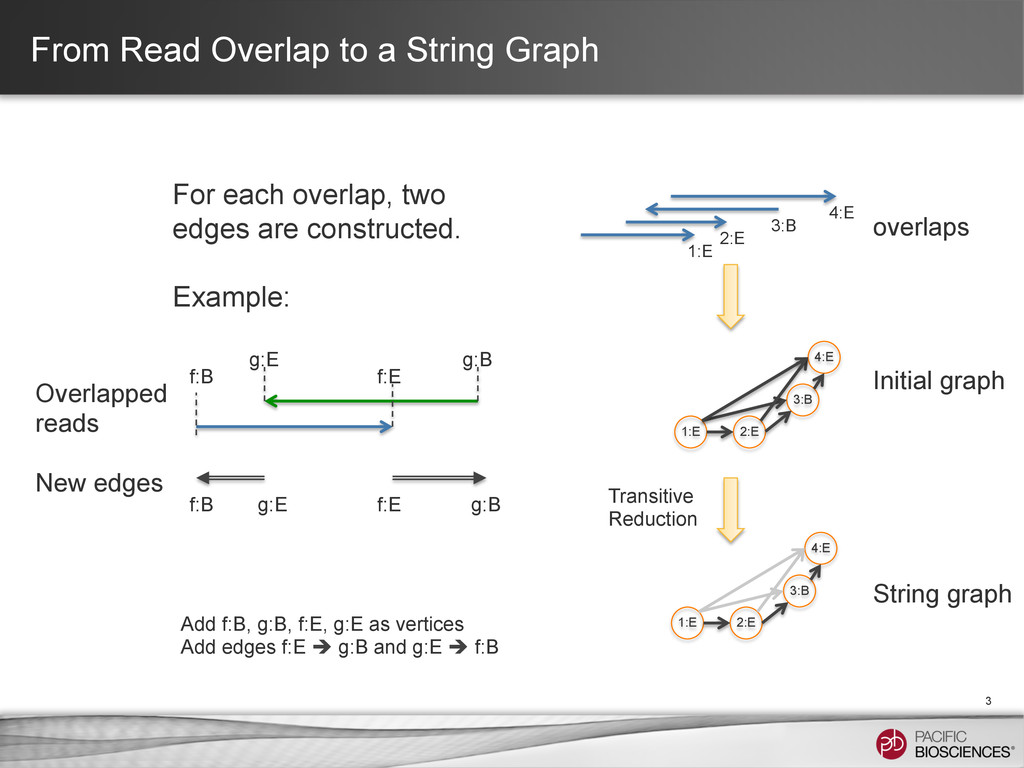
that models a genome • Nodes: – Particular positions (typically corresponding to the beginnings or endings of the read fragments) in the genome • Edges: – The sequence between the vertices • Any string from a path spell out a possible assembly from the reads Genome String Graph R1 R1 R2 R2 R2 R3 R3 e1 e2 e3 e4 e5 e1 e2 e3 e4 e5
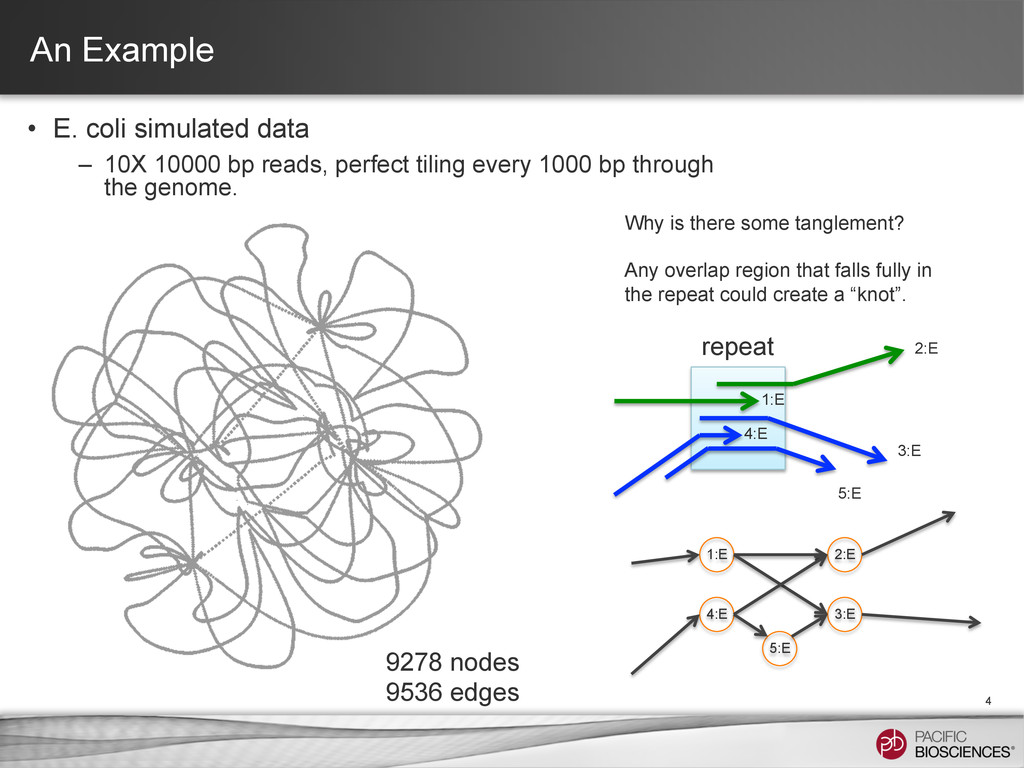
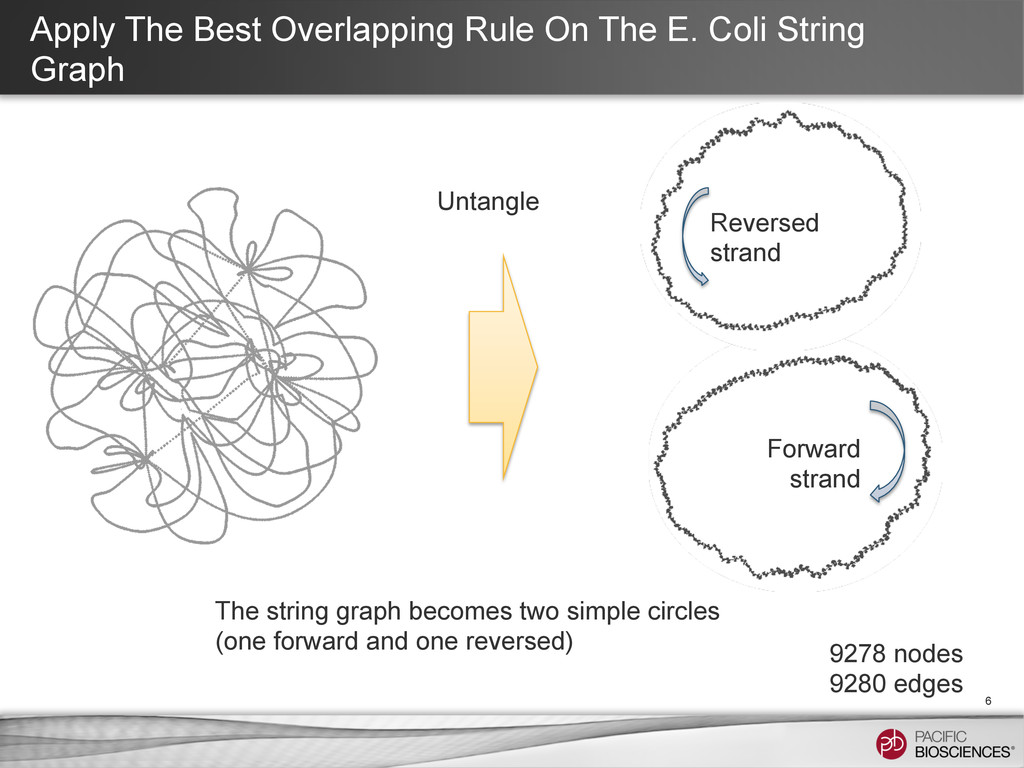
bp reads, perfect tiling every 1000 bp through the genome. 4 9278 nodes 9536 edges Why is there some tanglement? Any overlap region that falls fully in the repeat could create a “knot”. 1:E 2:E 3:E 4:E 5:E 1:E 2:E 3:E 4:E 5:E repeat
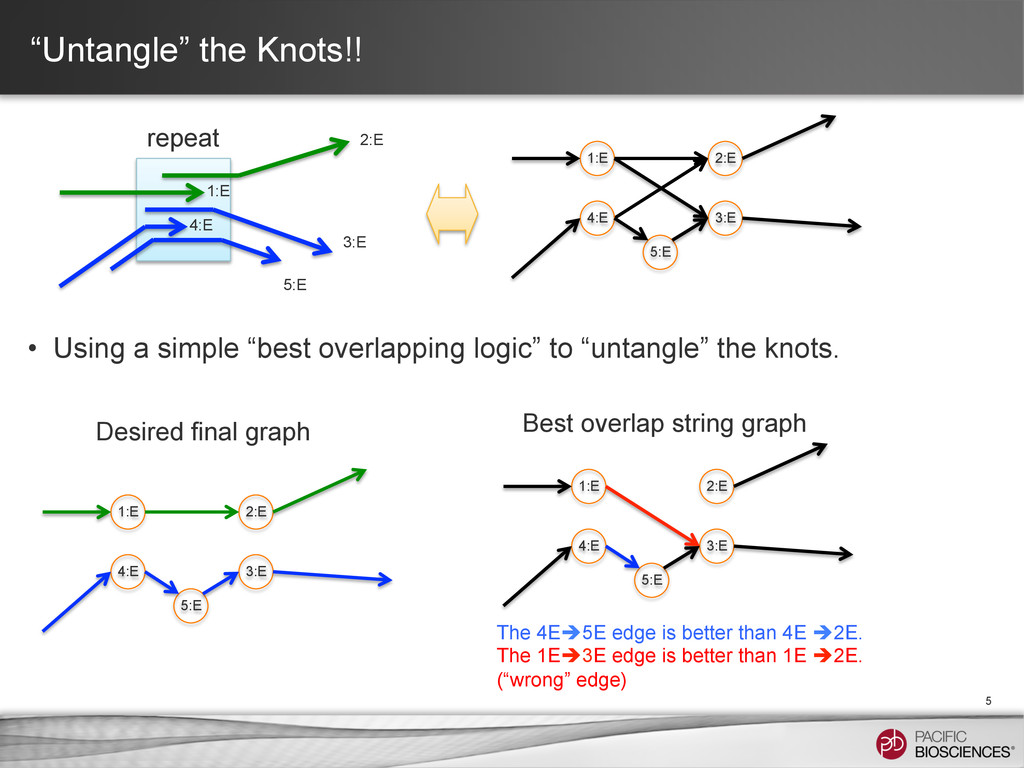
to “untangle” the knots. 5 1:E 2:E 3:E 1:E 2:E 3:E 4:E 4:E 5:E 5:E The 4Eè5E edge is better than 4E è2E. The 1Eè3E edge is better than 1E è2E. (“wrong” edge) 1:E 2:E 3:E 4:E 5:E Best overlap string graph 1:E 2:E 3:E 4:E 5:E Desired final graph repeat
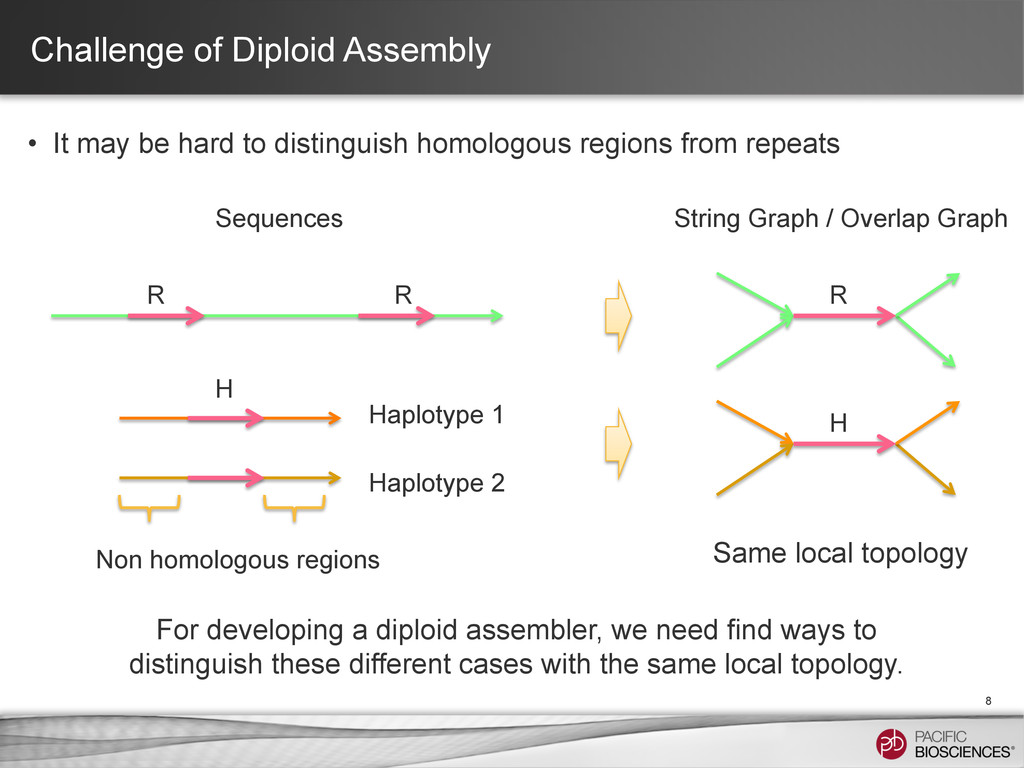
distinguish homologous regions from repeats 8 R R Haplotype 1 Haplotype 2 Non homologous regions Sequences String Graph / Overlap Graph R H H For developing a diploid assembler, we need find ways to distinguish these different cases with the same local topology. Same local topology
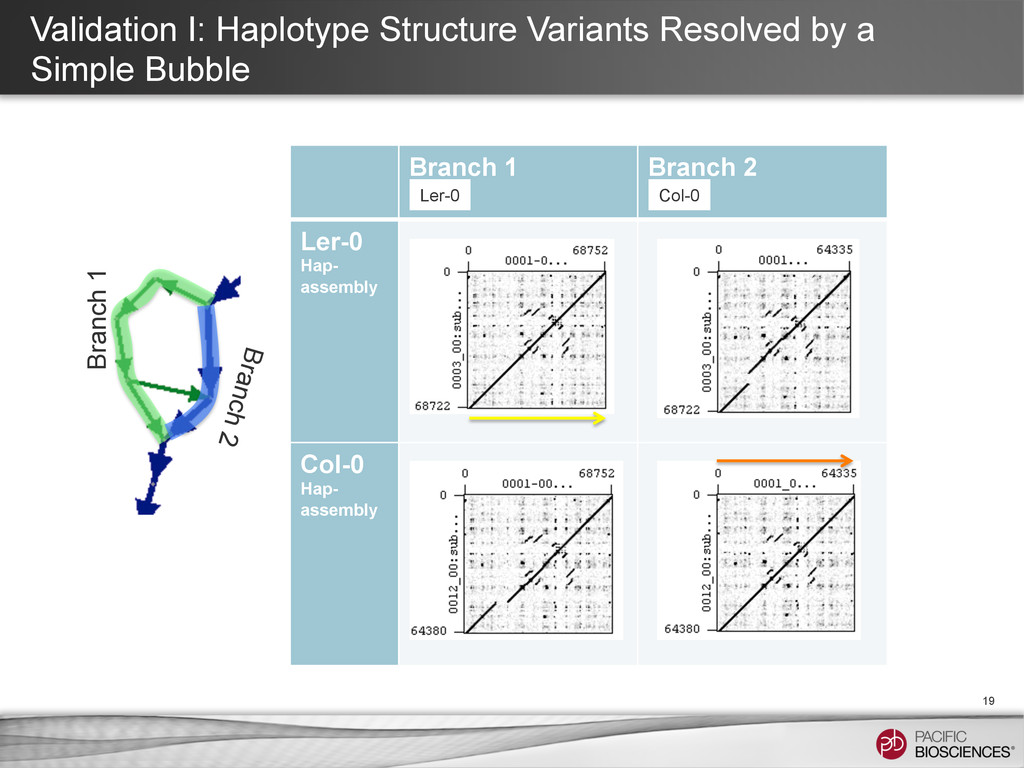
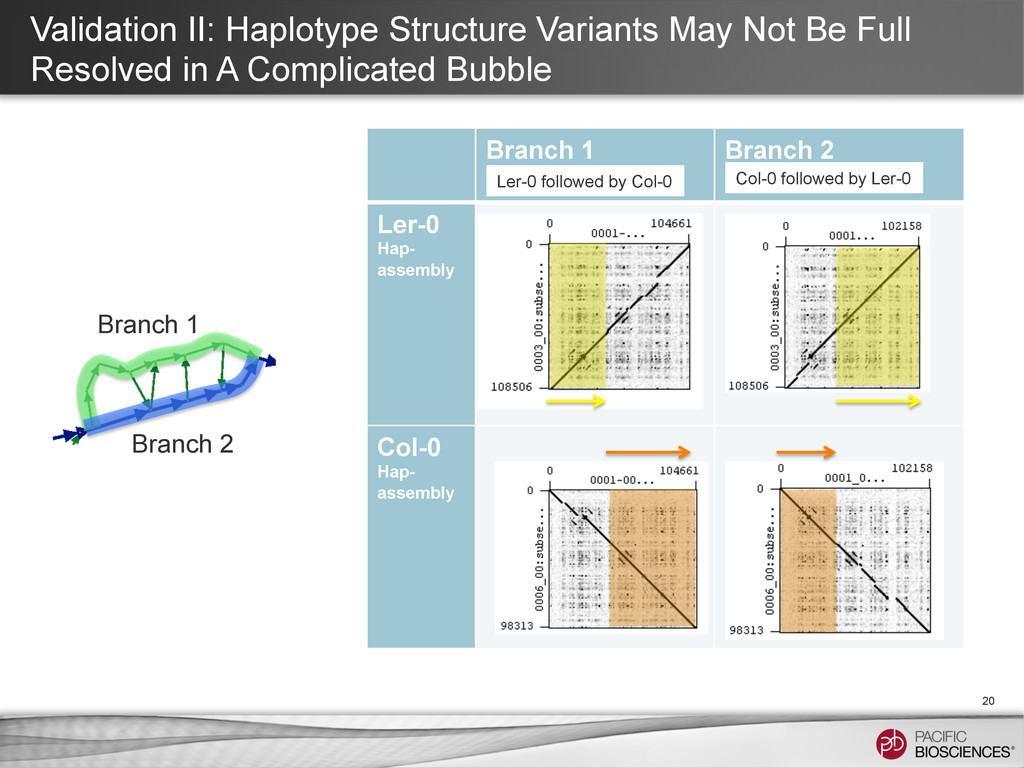
4:E 5:E 6:E 7:E 8:E SV or Error 1 2 3 4 5 6 7 8 With long reads, the string graph may have a quasi linear structure with bubbles induced by variations between haplotypes. Hopefully, with longer read length, most difference between two haplotypes appears as such simple bubble.
caused by SV between homologous copies Branching point caused by repeats An unitig graph from Ler-0 + Col-0 data The graph “diameter” ~ 12 M bp Mean edge size=17.4 k bp
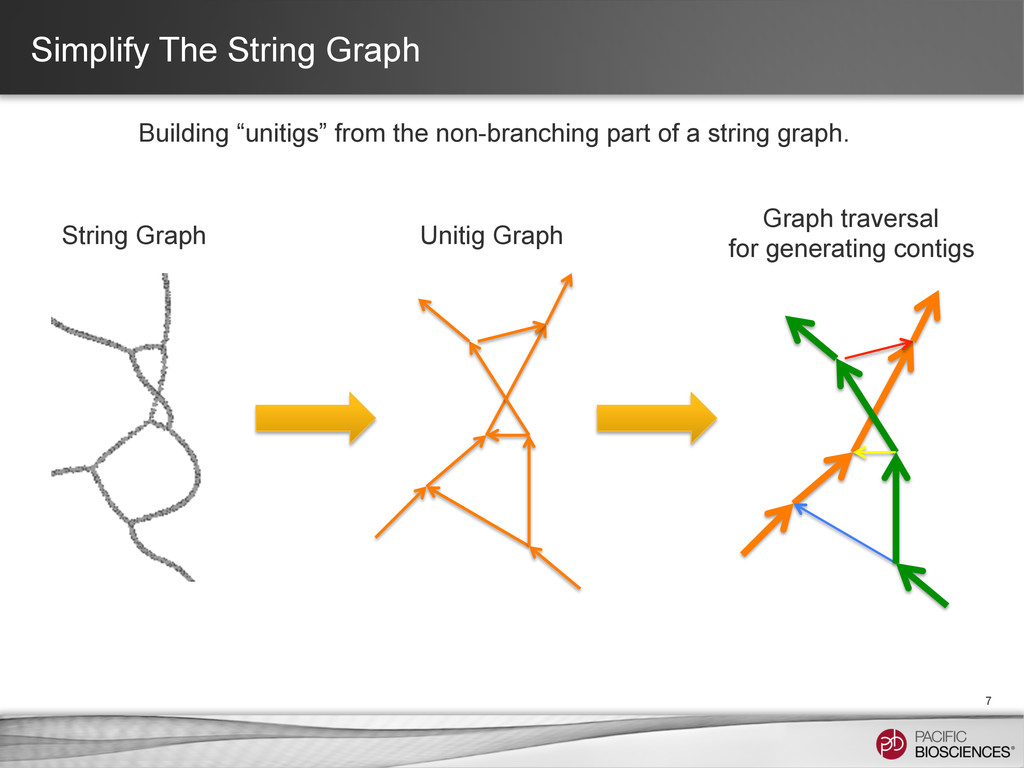
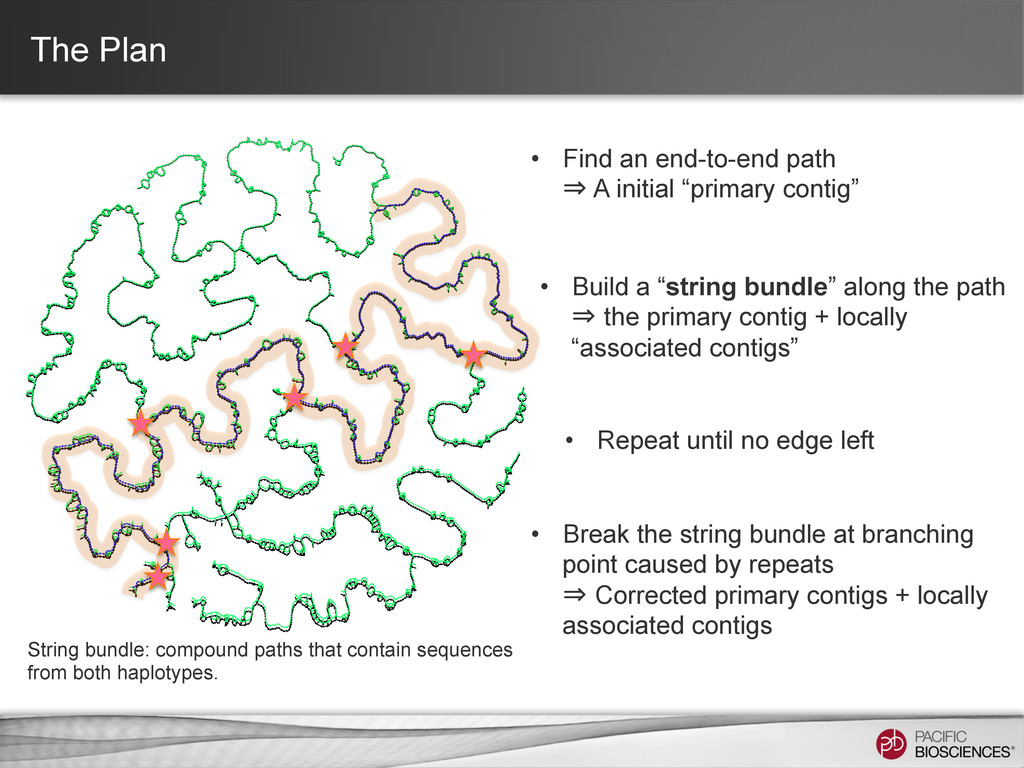
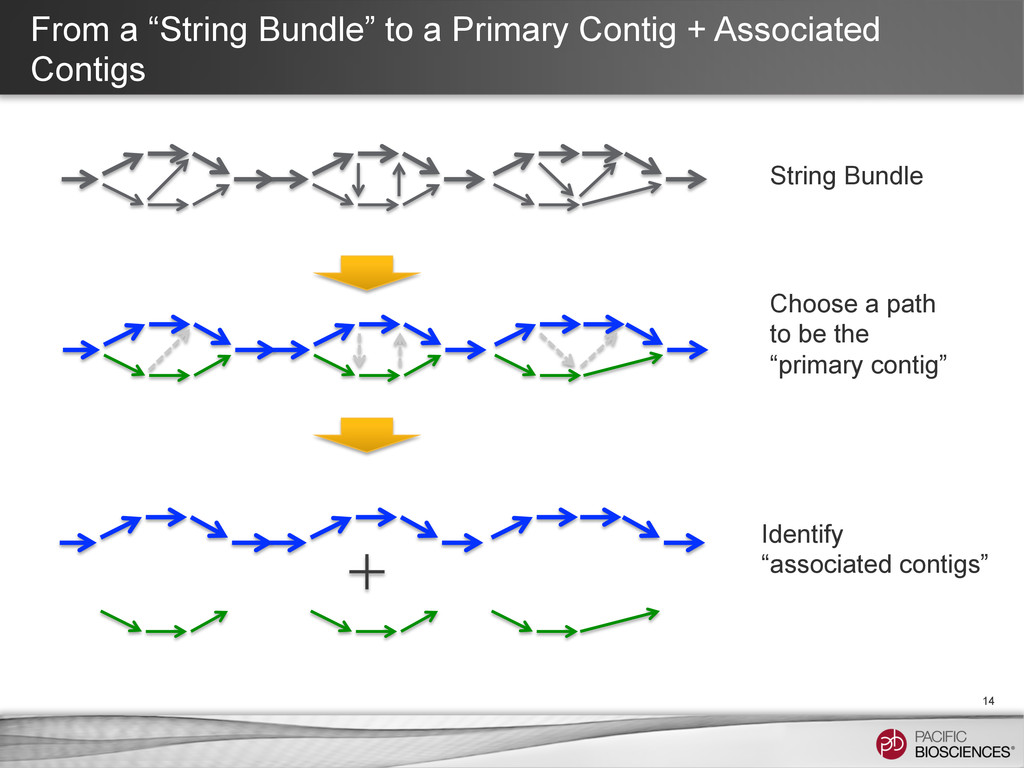
⇒ the primary contig + locally “associated contigs” • Break the string bundle at branching point caused by repeats ⇒ Corrected primary contigs + locally associated contigs • Find an end-to-end path ⇒ A initial “primary contig” • Repeat until no edge left String bundle: compound paths that contain sequences from both haplotypes.
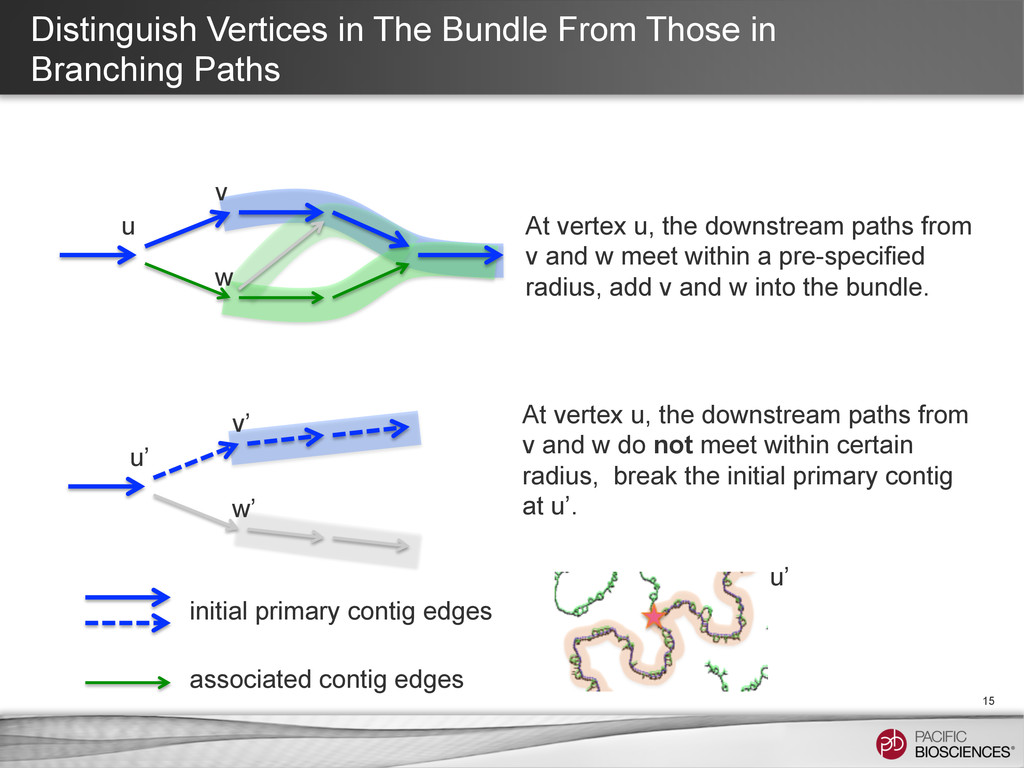
15 u’ u v’ w’ v w At vertex u, the downstream paths from v and w meet within a pre-specified radius, add v and w into the bundle. At vertex u, the downstream paths from v and w do not meet within certain radius, break the initial primary contig at u’. initial primary contig edges associated contig edges u’
Stats #Seqs 512 Max 10.2 Mbp Total 120 Mbp n50 6.2 Mbp From tick mark to tick mark is a contig. TAIR10, Col-0 Reference Assembled Contigs un-used edges in the original string graph
Stats #Seqs 1085 Max 9.4 Mbp Total 127 Mbp n50 2.8 Mbp TAIR10, Col-0 Reference Assembled Contigs From tick mark to tick mark is a contig. Full Contigs Stats #Seqs 2483 Max 12.5 Mbp (un-corrected) Total 177 Mbp n50 2.8 Mbp
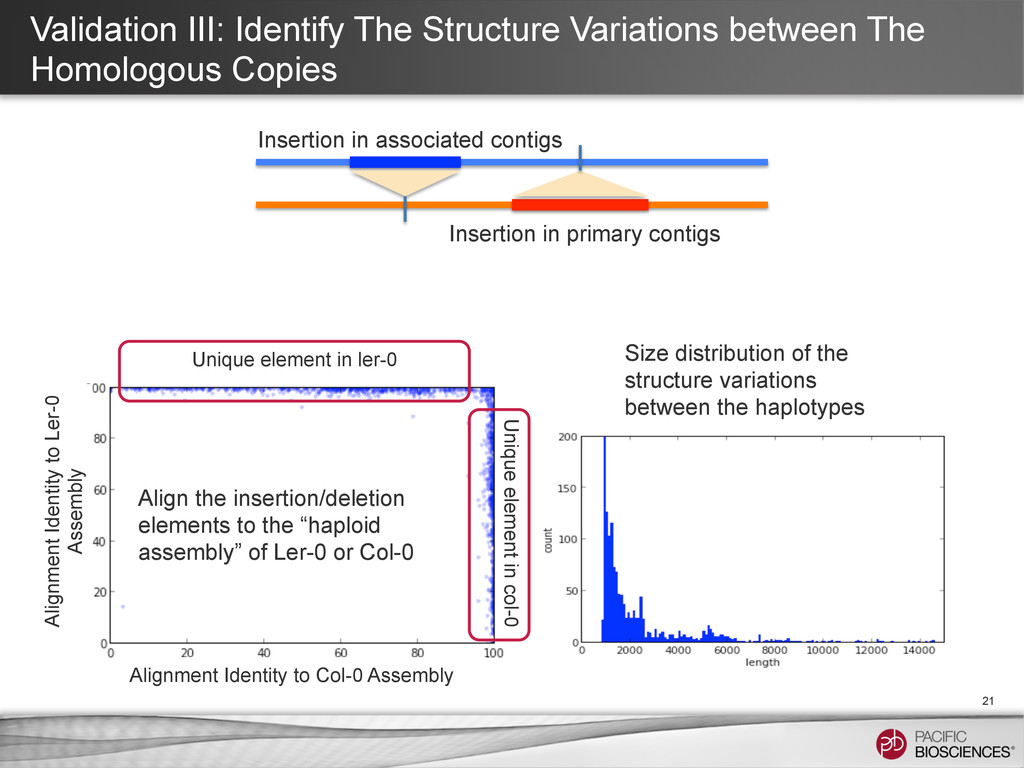
21 Insertion in associated contigs Insertion in primary contigs Align the insertion/deletion elements to the “haploid assembly” of Ler-0 or Col-0 Unique element in ler-0 Unique element in col-0 Size distribution of the structure variations between the haplotypes Alignment Identity to Ler-0 Assembly Alignment Identity to Col-0 Assembly
• With enough long read data, starting assembly from reads > 10kb to reveals the diploid structure as quasi-linear chains (string bundles) in the string graph. • Successfully assemble “diploid”-like long read data with heuristics. N50 > 5 Mb for haploid and N50 > 2.5 Mb for diploid. • Next: – Diploid / polyploid graph traversing problem: from heuristics to more rigorous theoretical framework – Generate diploid consensus: need an efficient aligner that can align long reads to string graph directly – Phasing: combining SV discoveries and SNP calling to “unzip” the bubbles – “Serialize the graph”: FASTG as output? – More testing cases: − Real biological diploid genomes − Other diploid genome might have different structure • Datasets: https://github.com/PacificBiosciences/DevNet/wiki/Datasets just search “PacBio Dataset” 22
team effort to bring useful data for the community. • Col-0 DNA Sample – Joe Ecker and Chongyun Lou (HHMI & Salk Institute) • For several important things I learned about assembly tools/algorithm through social networks: – Michael Schatz (@mike_schatz, CSHL), Adam Phillippy (@aphillippy , NBACC) • Blasr, a long read aligner: – Mark Chaisson (University of Washington, Eichler’s lab) 23
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}