Upgrade to Pro
— share decks privately, control downloads, hide ads and more …
Speaker Deck
Features
Speaker Deck
PRO
Sign in
Sign up for free
Search
Search
Introduction to R for Genomic Data Analysis
Search
onouyek
September 25, 2020
Programming
430
0
Share
Embed
Copy iframe code
Copy JS code
Copy link
Start on current slide
Introduction to R for Genomic Data Analysis
【第2回】ゼロから始めるゲノム解析(R編)
onouyek
September 25, 2020
More Decks by onouyek
See All by onouyek
Finding Open Reading Frames
onouyek
0
400
Inferring mRNA from Protein: Products and Reductions of Lists
onouyek
0
380
Finding the Longest Shared Subsequence: Finding K-mers, Writing Functions, and Using Binary Search
onouyek
0
300
Find a Motif in DNA: Exploring Sequence Similarity
onouyek
0
350
Finding the Hamming Distance: Counting Point Mutations
onouyek
0
470
Creating the Fibonacci Sequence: Writing, Testing, and Benchmarking Algorithms
onouyek
1
500
Transcribing DNA into mRNA: Mutating Strings, Reading and Writing Files
onouyek
0
560
DNA methylation analysis using bisulfite sequencing data
onouyek
0
920
Operations on Genomic Intervals and Genome Arithmetic
onouyek
0
370
Other Decks in Programming
See All in Programming
OS アップデート対応の取り組み方がもっと共有されてほしい
andpad
0
110
自作OSでスライド発表する
uyuki234
1
3.7k
エンジニアと一緒にテストコードの設計と実装を改善した話
mototakatsu
0
250
ローカルLLMでどこまでコードが書けるか -縮小版 / How much code can be written on a local LLM Shortened
kishida
2
180
The Bowling Game - From Imperative to Functional Programming - Part 1
philipschwarz
PRO
0
280
Dataformのリポジトリを立ち上げるときにまずやること / dataform-day0-2026
snhryt
0
210
SREは、MCPとSRE Agentをこう使え!
kazumax55
0
150
技術記事、 専門家としてのプログラマ、 言語化
mizchi
14
7.3k
Generative UI & AI-Assistants for Your Angular Solutions
manfredsteyer
PRO
0
140
「なぜそう決めたのか」を残し続ける仕組み ― Notion AI カスタムエージェント × Slack連携による設計判断の自動記録 - NIKKEI Tech Talk #47
niftycorp
PRO
0
250
SREの積み重ねがAI駆動開発のガードレールになった ― 7つの実践/SRE Guardrails The 7
tomoyakitaura
7
2.8k
その問い、本当に正しいですか?AI時代のエンジニアに必要な哲学と認知科学 / ai-philosophy-cognitive-science
minodriven
14
6.7k
Featured
See All Featured
Have SEOs Ruined the Internet? - User Awareness of SEO in 2025
akashhashmi
0
380
Producing Creativity
orderedlist
PRO
348
40k
Creating an realtime collaboration tool: Agile Flush - .NET Oxford
marcduiker
35
2.5k
Designing Powerful Visuals for Engaging Learning
tmiket
1
440
AI: The stuff that nobody shows you
jnunemaker
PRO
8
770
Making Projects Easy
brettharned
120
6.7k
The World Runs on Bad Software
bkeepers
PRO
72
12k
Building an army of robots
kneath
306
46k
WCS-LA-2024
lcolladotor
0
670
Fight the Zombie Pattern Library - RWD Summit 2016
marcelosomers
234
17k
Neural Spatial Audio Processing for Sound Field Analysis and Control
skoyamalab
0
370
16th Malabo Montpellier Forum Presentation
akademiya2063
PRO
0
170
Transcript
ゲノム解析のためのR入門 【第2回】ゼロから始めるゲノム解析 2020 年 9 月 25 日 onouyek@Bioinformatics Tokyo
目的 データ分析の手順を理解してもらい、ゲノム解析に必要なRプログラミングの基本を 習得します。
目次 • データ分析の流れ • パッケージのインストール • 算術演算 • データ構造 •
データ型 • データの読み取りと書き込み • プロット • 関数と制御構造
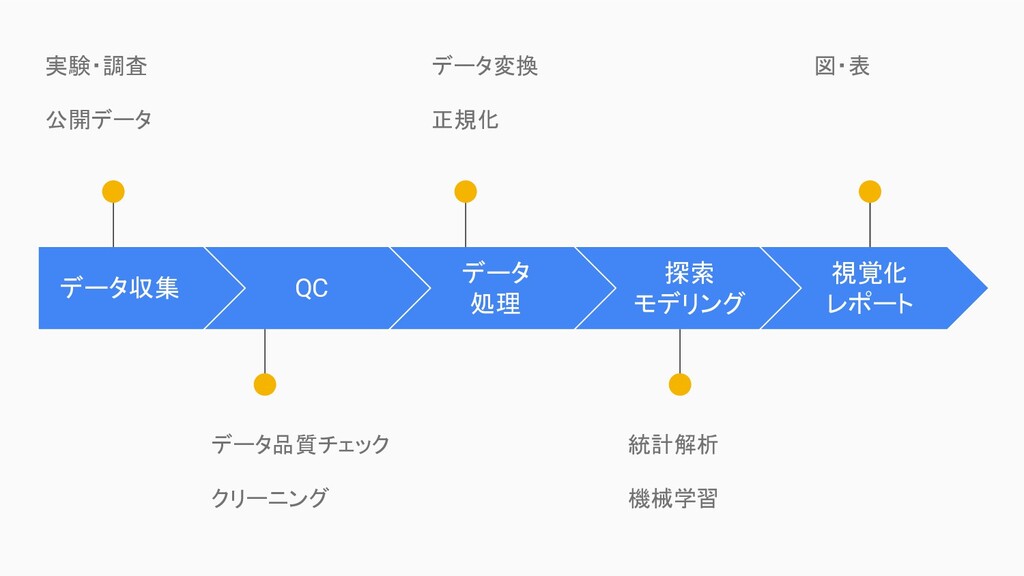
データ分析の流れ
データ収集 実験・調査 公開データ QC データ品質チェック クリーニング データ 処理 データ変換 正規化
探索 モデリング 統計解析 機械学習 視覚化 レポート 図・表
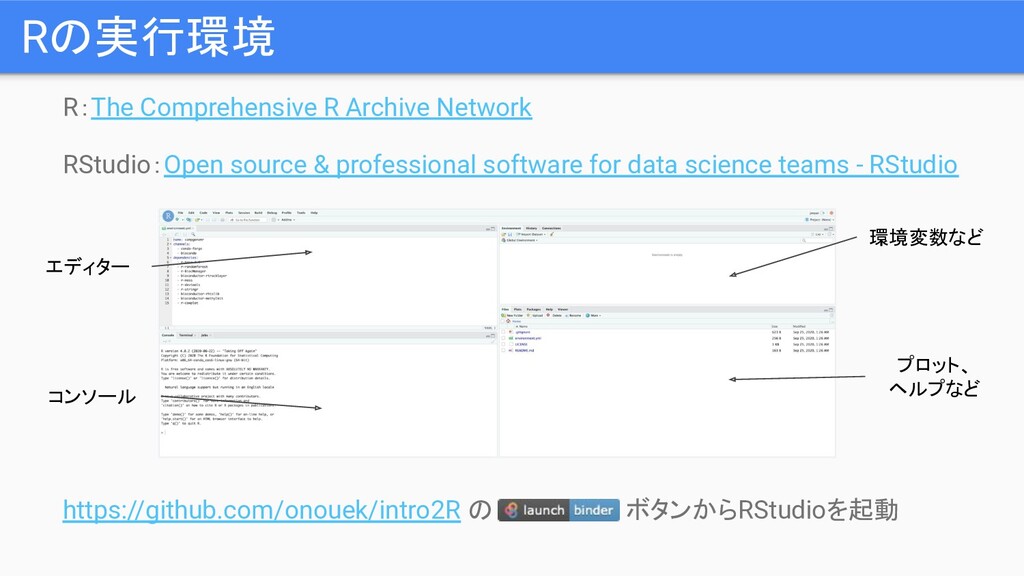
R:The Comprehensive R Archive Network RStudio:Open source & professional software
for data science teams - RStudio https://github.com/onouek/intro2R の ボタンからRStudioを起動 Rの実行環境 エディター コンソール 環境変数など プロット、 ヘルプなど
パッケージのインストール (CRAN, Bioconductor) パッケージ:ベースRへのアドオン CRAN:Rのメインパッケージリポジトリ Bioconductor:計算生物学に関連するパッケージ専用のパッケージリポジトリ # #で始まる行はコメント行 # CRANの"randomForest"パッケージのインストール
install.packages("randomForest") # Bioconductorのパッケージインストーラー”BiocManager”のインストール install.packages("BiocManager") # Bioconductorの"rtracklayer"のインストール BiocManager::install("rtracklayer")
パッケージのインストール (GitHub, source file) # devtoolsパッケージを使ってGitHubから"compgenomr/compGenomRData"パッケージのインストール library(devtools) install_github("compgenomr/compGenomRData") # パッケージのsource
fileをダウンロード download.file( "https://github.com/tidyverse/stringr/archive/v1.4.0.tar.gz", destfile="v1.4.0.tar.gz") # source fileからインストール install.packages("v1.4.0.tar.gz",repos=NULL,type="source") # source fileの削除 unlink("v1.4.0.tar.gz")
パッケージと関数のヘルプの出し方 # “MASS”パッケージの関数のリスト library(MASS) ls("package:MASS") # R環境にあるオブジェクトのリスト ls() # hist()のヘルプの取得
?hist help("hist") # "hist"にマッチするヘルプページの検索 help.search("hist") ??hist
算術演算 2 + 3 # 足し算 3 * 5 #
掛け算 2 + 3 * 5 # 順番に注意! 5^2 # 累乗 3/2 # 割り算 log(10) # 自然対数 sqrt(16) # 平方根 abs(3-7) # 絶対値 exp(2) # 指数関数 pi # 円周率
データ構造
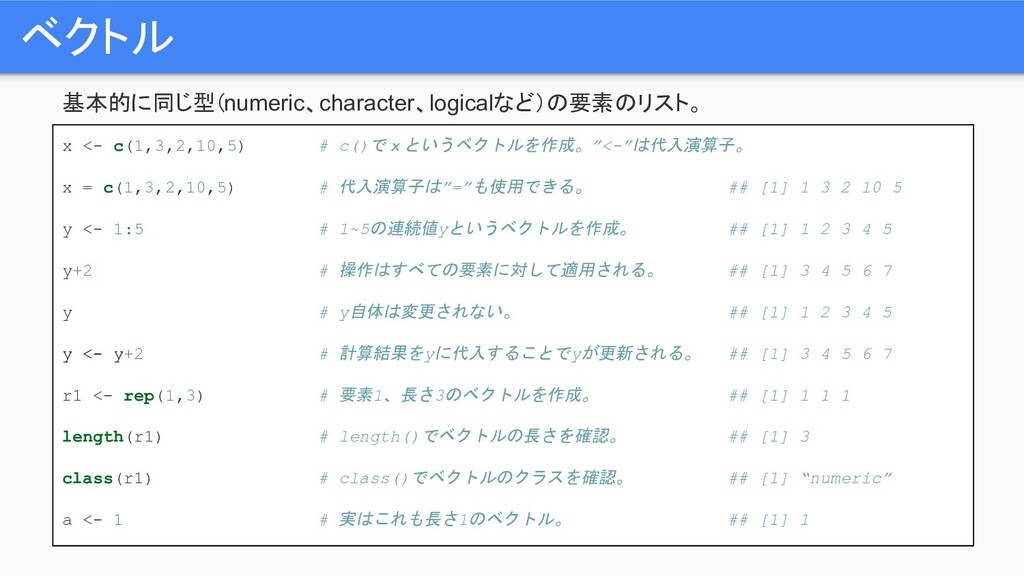
ベクトル 基本的に同じ型(numeric、character、logicalなど)の要素のリスト。 x <- c(1,3,2,10,5) # c()でxというベクトルを作成。”<-”は代入演算子。 x = c(1,3,2,10,5)
# 代入演算子は”=”も使用できる。 ## [1] 1 3 2 10 5 y <- 1:5 # 1~5の連続値yというベクトルを作成。 ## [1] 1 2 3 4 5 y+2 # 操作はすべての要素に対して適用される。 ## [1] 3 4 5 6 7 y # y自体は変更されない。 ## [1] 1 2 3 4 5 y <- y+2 # 計算結果をyに代入することでyが更新される。 ## [1] 3 4 5 6 7 r1 <- rep(1,3) # 要素1、長さ3のベクトルを作成。 ## [1] 1 1 1 length(r1) # length()でベクトルの長さを確認。 ## [1] 3 class(r1) # class()でベクトルのクラスを確認。 ## [1] “numeric” a <- 1 # 実はこれも長さ1のベクトル。 ## [1] 1
行列 行と列の数値配列。各行または列がベクトル。 x <- c(1,2,3,4) y <- c(4,5,6,7) m1 <-
cbind(x,y) # cbind()で列で結合 t(m1) # t()で転置 dim(m1) # dim()で次元を確認 m2 <- matrix(c(1,3,2,5,-1,2,2,3,9),nrow=3) # matrix()で要素ベクトルと行数を指定 m2 ## [,1] [,2] [,3] ## [1,] 1 5 2 ## [2,] 3 -1 3 ## [3,] 2 2 9
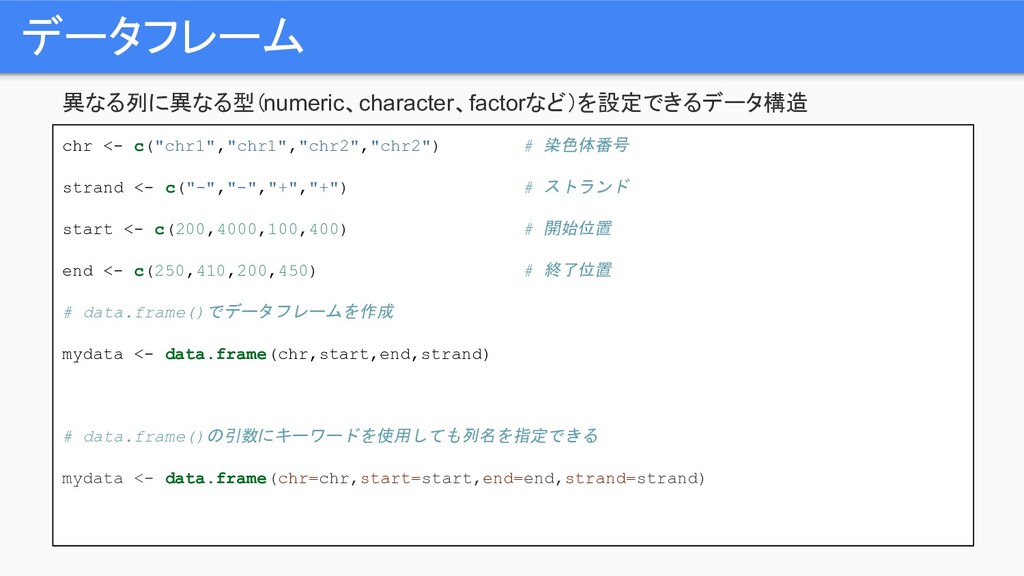
データフレーム 異なる列に異なる型(numeric、character、factorなど)を設定できるデータ構造 chr <- c("chr1","chr1","chr2","chr2") # 染色体番号 strand <- c("-","-","+","+")
# ストランド start <- c(200,4000,100,400) # 開始位置 end <- c(250,410,200,450) # 終了位置 # data.frame()でデータフレームを作成 mydata <- data.frame(chr,start,end,strand) # data.frame()の引数にキーワードを使用しても列名を指定できる mydata <- data.frame(chr=chr,start=start,end=end,strand=strand)
データフレームの要素の抽出 列番号または名前で特定の列を抽出したり、行番号で特定の行を抽出したりできる。 mydata[,2:4] # 列番号2,3,4の列を抽出 mydata[,c("chr","start")] # 列名がchrとstartの列を抽出 mydata$start #
列名がstartの列を抽出 mydata[c(1,3),] # 行番号1,3の行を抽出 mydata[mydata$start>400,] # start列が400以上の行を抽出
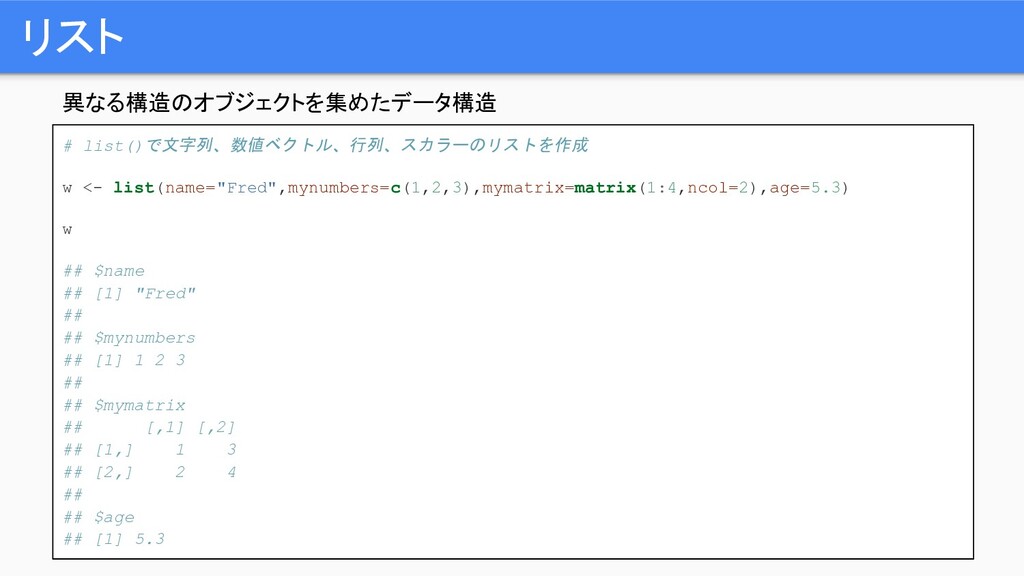
リスト 異なる構造のオブジェクトを集めたデータ構造 # list()で文字列、数値ベクトル、行列、スカラーのリストを作成 w <- list(name="Fred",mynumbers=c(1,2,3),mymatrix=matrix(1:4,ncol=2),age=5.3) w ## $name
## [1] "Fred" ## ## $mynumbers ## [1] 1 2 3 ## ## $mymatrix ## [,1] [,2] ## [1,] 1 3 ## [2,] 2 4 ## ## $age ## [1] 5.3
リストの要素の抽出 位置または名前を二重括弧で指定して抽出できる。 # 位置(3番目)を指定してオブジェクトを抽出 w[[3]] ## [,1] [,2] ## [1,]
1 3 ## [2,] 2 4 # 名前(mynumbers)を指定してオブジェクトを抽出 w[["mynumbers"]] ## [1] 1 2 3 # 名前(age)を$で指定してオブジェクトを抽出 w$age ## [1] 5.3
ファクター カテゴリーデータを格納するためのデータ構造 ※data.frame()でデータフレームを作成した場合に、文字列はデフォルトでファクターとして格納される。 features=c("promoter","exon","intron") f.feat=factor(features) # factor()関数でf.featにカテゴリーデータを格納 f.feat ## [1]
promoter exon intron ## Levels: exon intron promoter
データ型 Rの4つのデータ型(numeric、logical、character、integer) # numericベクトルxを作成 x <- c(1,3,2,10,5) ## [1] 1
3 2 10 5 # logicalベクトルxを作成 x <- c(TRUE,FALSE,TRUE) ## [1] TRUE FALSE TRUE # characterベクトルxを作成 x <- c("sds","sd","as") ## [1] "sds" "sd" "as" class(x) ## [1] "character" # integerベクトルxを作成 x <- c(1L,2L,3L) ## [1] 1 2 3 class(x) ## [1] “integer”
データの読み取りと書き込み
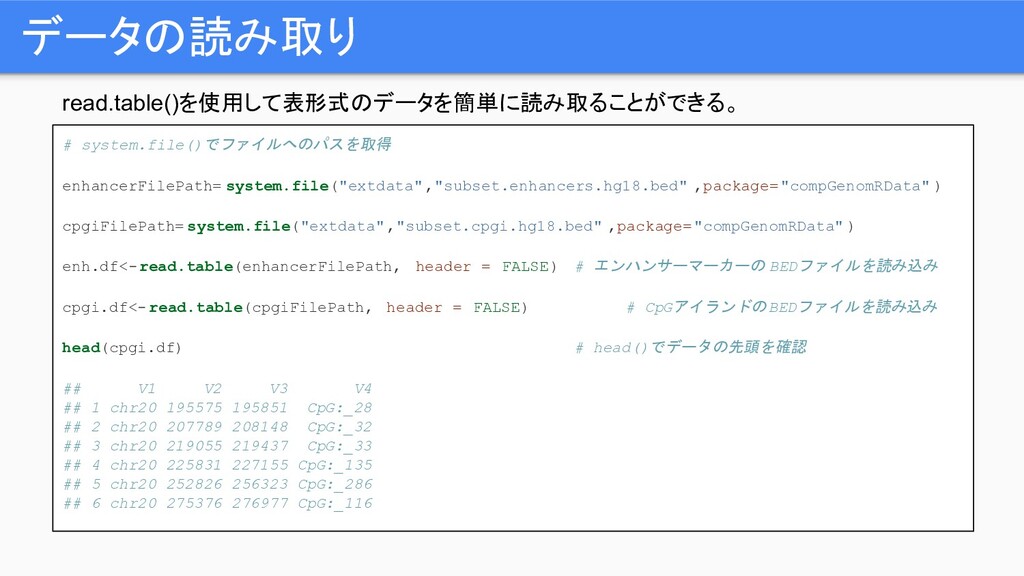
read.table()を使用して表形式のデータを簡単に読み取ることができる。 データの読み取り # system.file()でファイルへのパスを取得 enhancerFilePath= system.file("extdata","subset.enhancers.hg18.bed" ,package="compGenomRData" ) cpgiFilePath= system.file("extdata","subset.cpgi.hg18.bed"
,package="compGenomRData" ) enh.df<-read.table(enhancerFilePath, header = FALSE) # エンハンサーマーカーの BEDファイルを読み込み cpgi.df<-read.table(cpgiFilePath, header = FALSE) # CpGアイランドのBEDファイルを読み込み head(cpgi.df) # head()でデータの先頭を確認 ## V1 V2 V3 V4 ## 1 chr20 195575 195851 CpG:_28 ## 2 chr20 207789 208148 CpG:_32 ## 3 chr20 219055 219437 CpG:_33 ## 4 chr20 225831 227155 CpG:_135 ## 5 chr20 252826 256323 CpG:_286 ## 6 chr20 275376 276977 CpG:_116
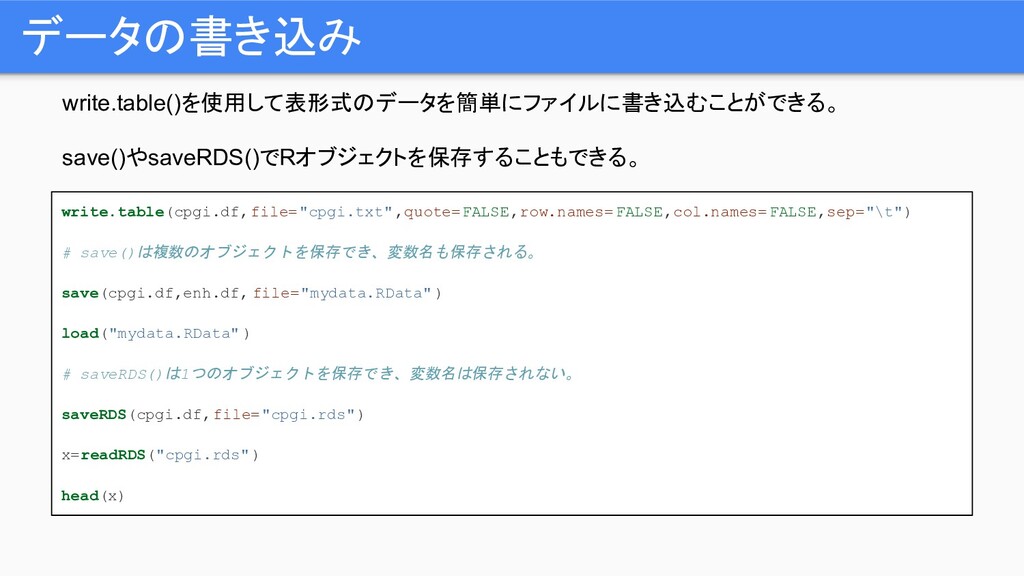
write.table()を使用して表形式のデータを簡単にファイルに書き込むことができる。 save()やsaveRDS()でRオブジェクトを保存することもできる。 データの書き込み write.table(cpgi.df,file="cpgi.txt",quote=FALSE,row.names=FALSE,col.names=FALSE,sep="\t") # save()は複数のオブジェクトを保存でき、変数名も保存される。 save(cpgi.df,enh.df, file="mydata.RData" ) load("mydata.RData"
) # saveRDS()は1つのオブジェクトを保存でき、変数名は保存されない。 saveRDS(cpgi.df,file="cpgi.rds") x=readRDS("cpgi.rds") head(x)
プロット
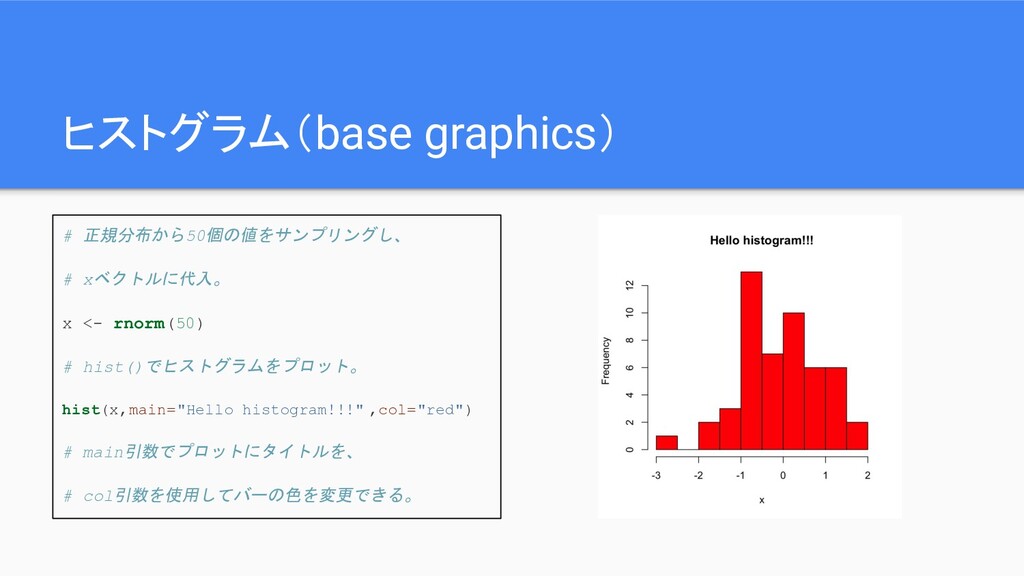
ヒストグラム(base graphics) # 正規分布から50個の値をサンプリングし、 # xベクトルに代入。 x <- rnorm(50) #
hist()でヒストグラムをプロット。 hist(x,main="Hello histogram!!!" ,col="red") # main引数でプロットにタイトルを、 # col引数を使用してバーの色を変更できる。
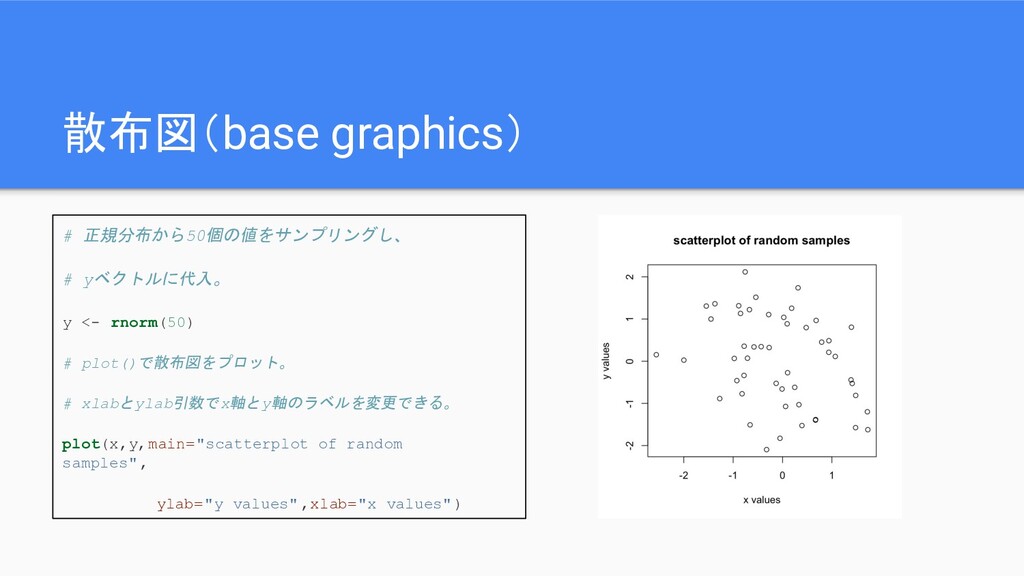
散布図(base graphics) # 正規分布から50個の値をサンプリングし、 # yベクトルに代入。 y <- rnorm(50) #
plot()で散布図をプロット。 # xlabとylab引数でx軸とy軸のラベルを変更できる。 plot(x,y,main="scatterplot of random samples", ylab="y values",xlab="x values")
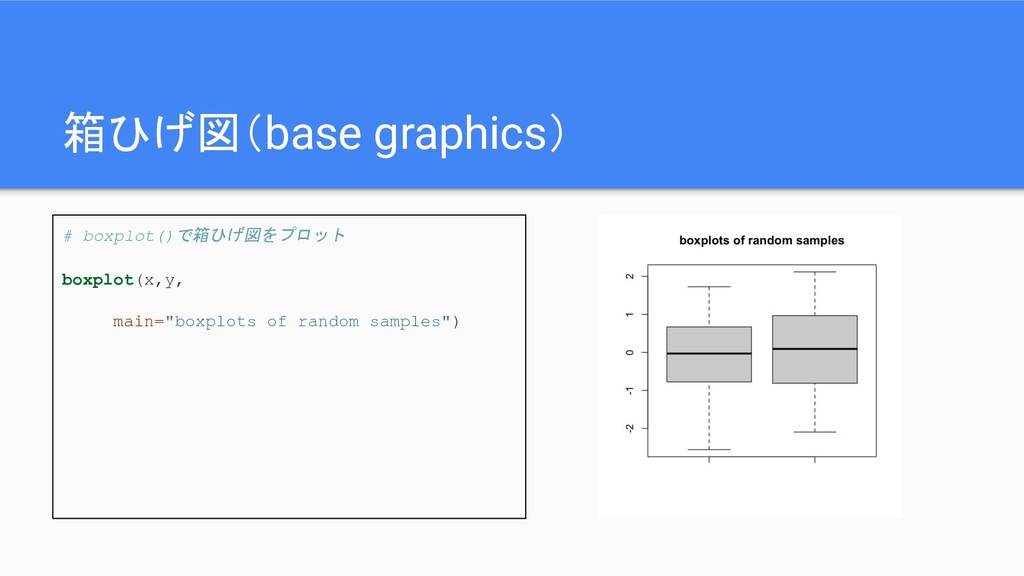
箱ひげ図(base graphics) # boxplot()で箱ひげ図をプロット boxplot(x,y, main="boxplots of random samples")
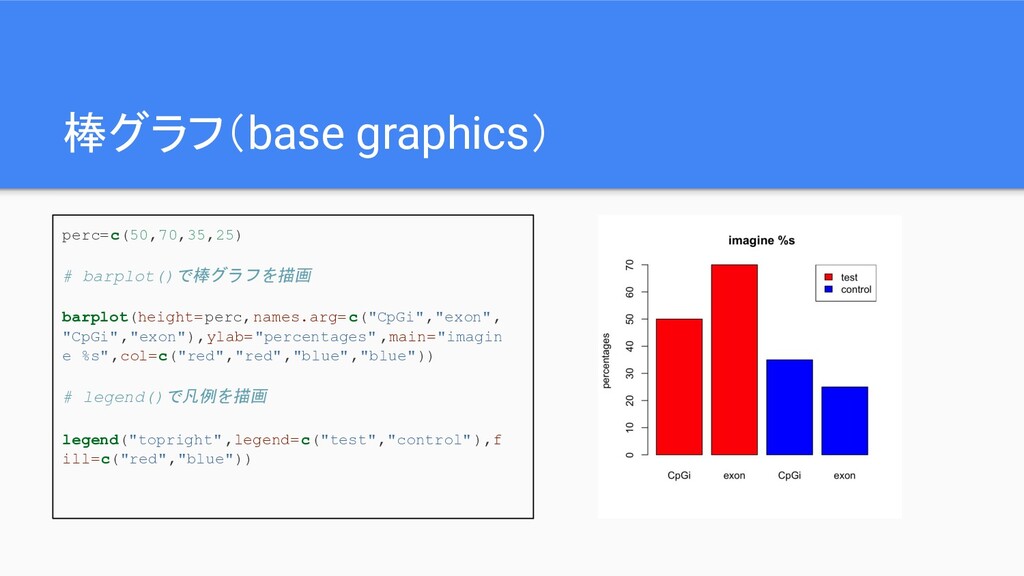
棒グラフ(base graphics) perc=c(50,70,35,25) # barplot()で棒グラフを描画 barplot(height=perc,names.arg=c("CpGi","exon", "CpGi","exon"),ylab="percentages" ,main="imagin e %s",col=c("red","red","blue","blue"))
# legend()で凡例を描画 legend("topright",legend=c("test","control"),f ill=c("red","blue"))
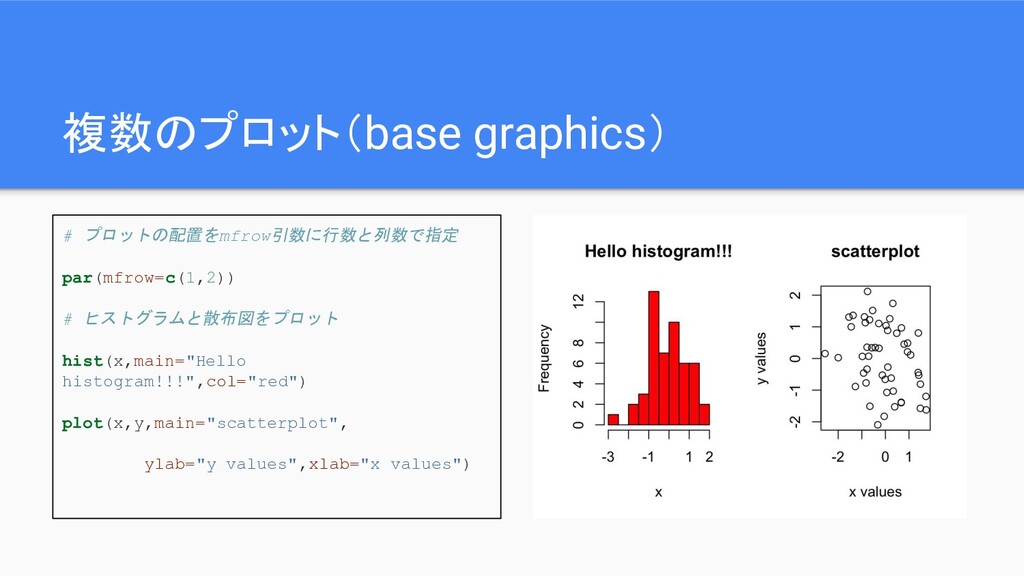
複数のプロット(base graphics) # プロットの配置をmfrow引数に行数と列数で指定 par(mfrow=c(1,2)) # ヒストグラムと散布図をプロット hist(x,main="Hello histogram!!!",col="red") plot(x,y,main="scatterplot",
ylab="y values",xlab="x values")
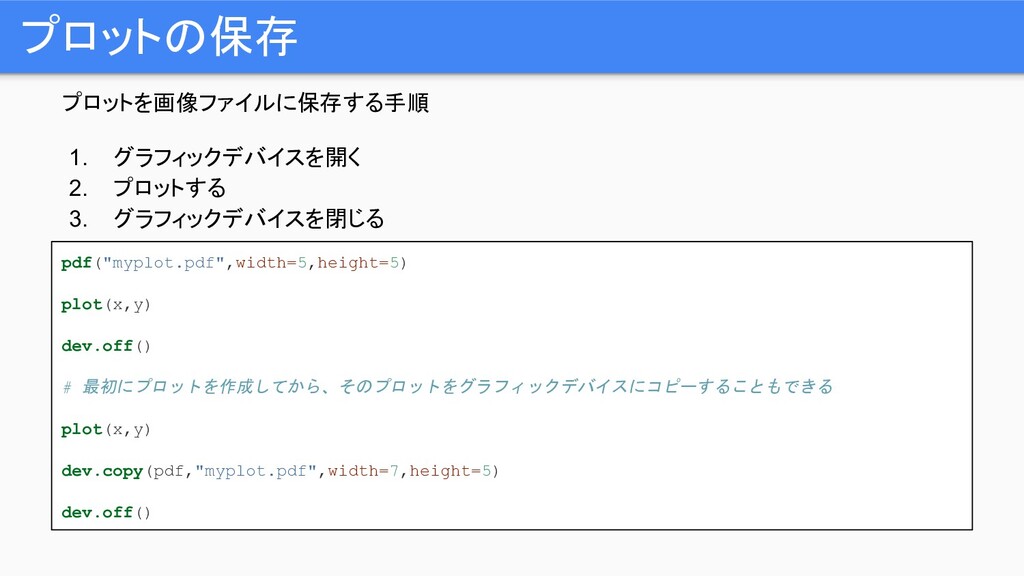
プロットを画像ファイルに保存する手順 1. グラフィックデバイスを開く 2. プロットする 3. グラフィックデバイスを閉じる プロットの保存 pdf("myplot.pdf",width=5,height=5) plot(x,y)
dev.off() # 最初にプロットを作成してから、そのプロットをグラフィックデバイスにコピーすることもできる plot(x,y) dev.copy(pdf,"myplot.pdf",width=7,height=5) dev.off()
散布図(ggplot2) library(ggplot2) myData=data.frame(col1=x,col2=y) # ggplot()の引数にmyDataデータフレームを指定 # aes()でmyDataのcol1とcol2の使用を指定 ggplot(myData, aes(x=col1, y=col2))
+ geom_point() # プロットの種類
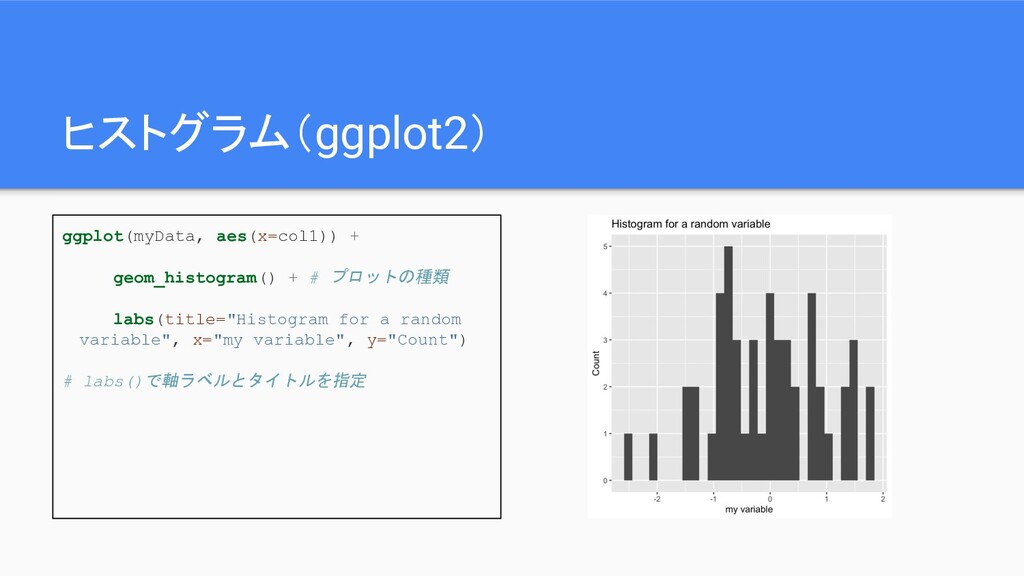
ヒストグラム(ggplot2) ggplot(myData, aes(x=col1)) + geom_histogram() + # プロットの種類 labs(title="Histogram for
a random variable", x="my variable", y="Count") # labs()で軸ラベルとタイトルを指定
箱ひげ図(ggplot2) # グループを追加する必要がある myData2=rbind( data.frame(values=x,group="x"), data.frame(values=y,group="y")) ggplot(myData2, aes(x=group,y=values)) + geom_boxplot() #
プロットの種類 # aes()でx軸にグループ、y軸に値を指定
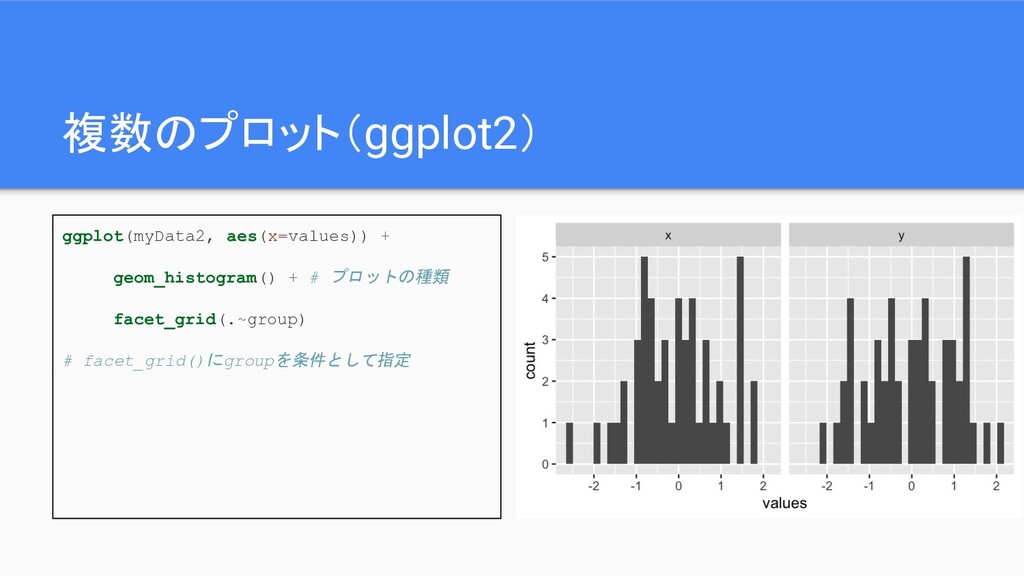
複数のプロット(ggplot2) ggplot(myData2, aes(x=values)) + geom_histogram() + # プロットの種類 facet_grid(.~group) #
facet_grid()にgroupを条件として指定
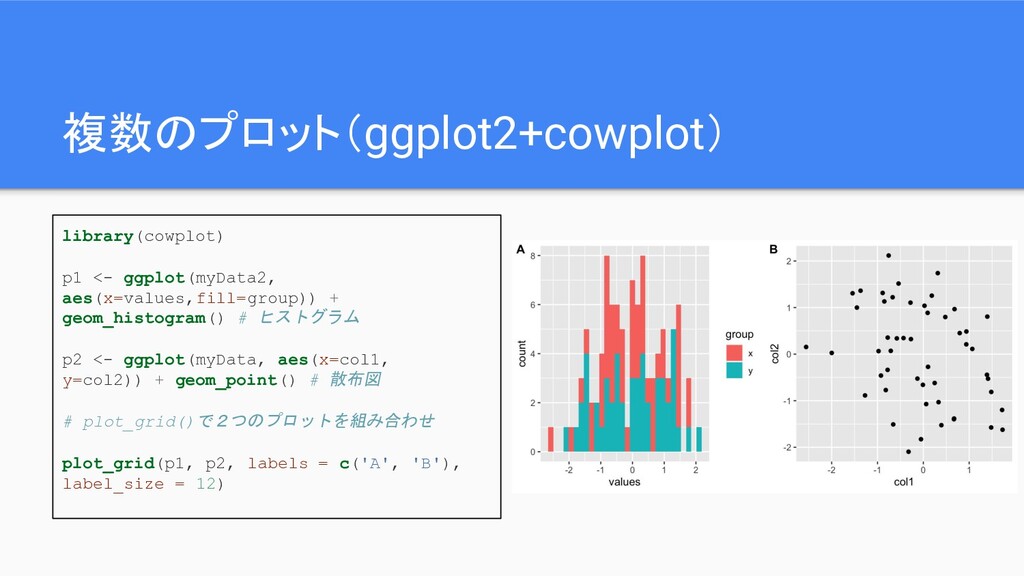
複数のプロット(ggplot2+cowplot) library(cowplot) p1 <- ggplot(myData2, aes(x=values,fill=group)) + geom_histogram() # ヒストグラム
p2 <- ggplot(myData, aes(x=col1, y=col2)) + geom_point() # 散布図 # plot_grid()で2つのプロットを組み合わせ plot_grid(p1, p2, labels = c('A', 'B'), label_size = 12)
ggplot2とtidyverse ggplot2はtidyverseという大きなエコシステムの一部です。 ggplot2: Elegant Graphics for Data Analysis:Hadley Wickhamによる無料の電子ブック Tidyverse.org:Tidyverseのパッケージとエコシステムについてのドキュメント
関数と制御構造
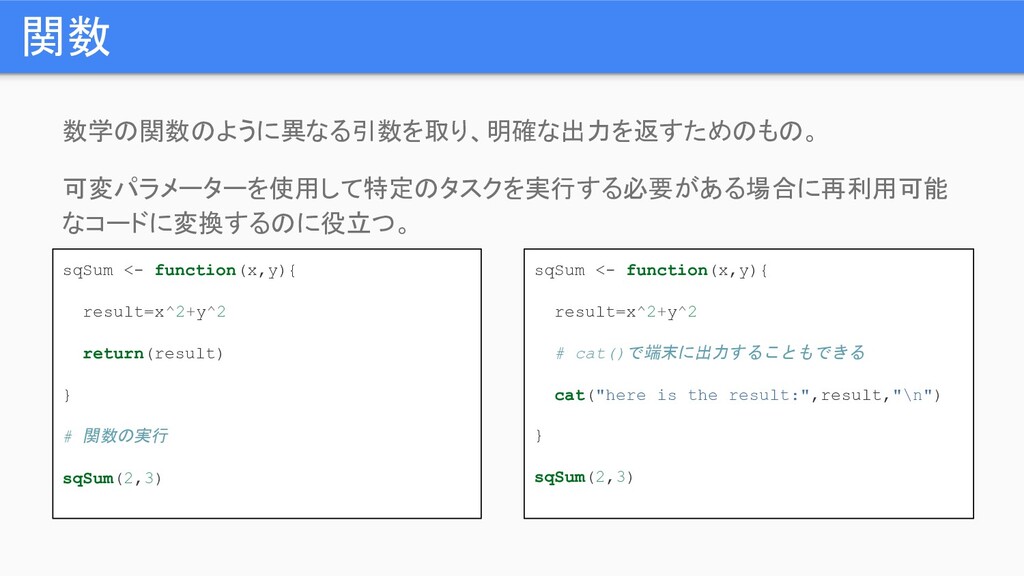
関数 数学の関数のように異なる引数を取り、明確な出力を返すためのもの。 可変パラメーターを使用して特定のタスクを実行する必要がある場合に再利用可能 なコードに変換するのに役立つ。 sqSum <- function(x,y){ result=x^2+y^2 return(result) }
# 関数の実行 sqSum(2,3) sqSum <- function(x,y){ result=x^2+y^2 # cat()で端末に出力することもできる cat("here is the result:",result,"\n") } sqSum(2,3)
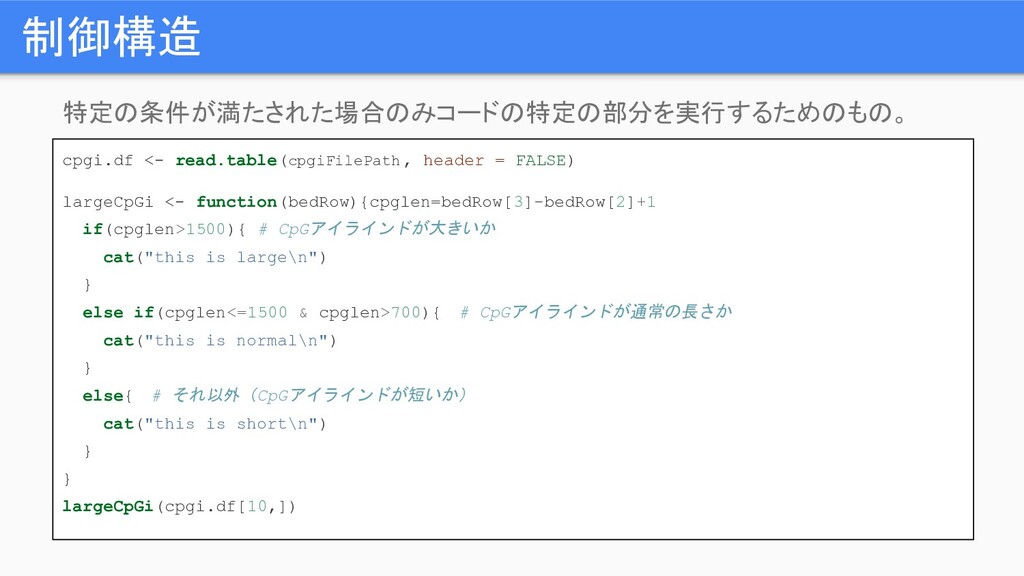
制御構造 特定の条件が満たされた場合のみコードの特定の部分を実行するためのもの。 cpgi.df <- read.table(cpgiFilePath , header = FALSE) largeCpGi
<- function(bedRow){cpglen=bedRow[3]-bedRow[2]+1 if(cpglen>1500){ # CpGアイラインドが大きいか cat("this is large\n") } else if(cpglen<=1500 & cpglen>700){ # CpGアイラインドが通常の長さか cat("this is normal\n") } else{ # それ以外(CpGアイラインドが短いか) cat("this is short\n") } } largeCpGi(cpgi.df[10,])
ループ 特定のタスクを繰り返したり、関数を複数回実行したりするためのもの。 for(i in 1:10){ # 繰り返す回数 cat("This is iteration")
print(i) } result=c() for(i in 1:100){ # CpGアイランドの長さを計算 len=cpgi.df[i,3]-cpgi.df[i,2]+1 # 結果をresultに追加 result=c(result,len) } head(result)
ループより効率的なapply()ファミリー関数 apply():データフレームまたは行列の各行または列に関数を適用できる。 lapply():リストまたはベクトルに関数を適用できる。 mat=cbind(c(3,0,3,3),c(3,0,0,0), c(3,0,0,3),c(1,1,0,0), c(1,1,1,0),c(1,1,1,0)) result <- apply(mat,1,sum)# 1:行に適用
result ## [1] 12 3 5 6 # 引数に関数を定義することもできる result <- apply(mat,1,function(x) sum(x))
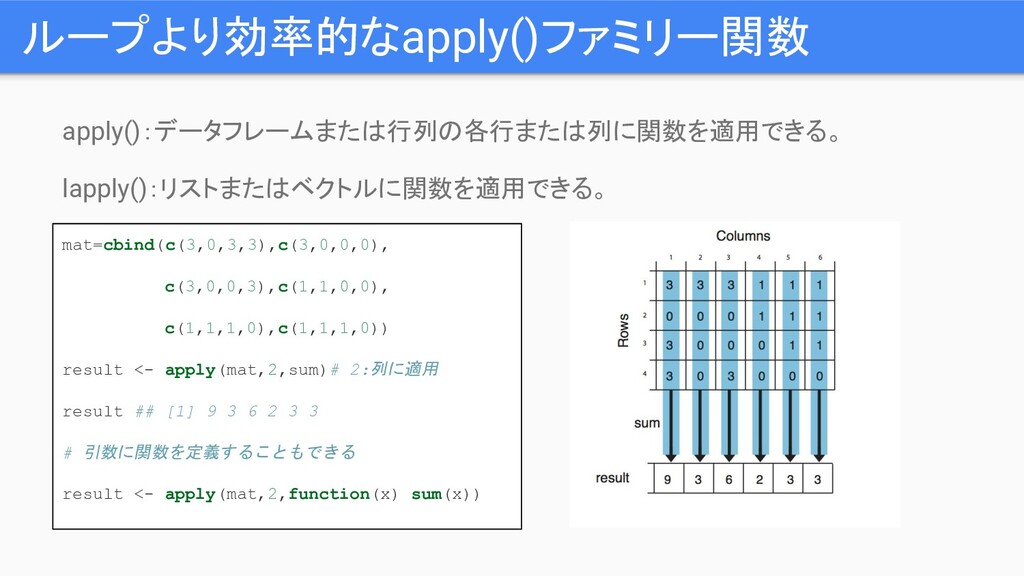
ループより効率的なapply()ファミリー関数 apply():データフレームまたは行列の各行または列に関数を適用できる。 lapply():リストまたはベクトルに関数を適用できる。 mat=cbind(c(3,0,3,3),c(3,0,0,0), c(3,0,0,3),c(1,1,0,0), c(1,1,1,0),c(1,1,1,0)) result <- apply(mat,2,sum)# 2:列に適用
result ## [1] 9 3 6 2 3 3 # 引数に関数を定義することもできる result <- apply(mat,2,function(x) sum(x))
ループより効率的なapply()ファミリー関数 mapply():ベクトル/リストの複数のセットに関数を適用できる。 Xs=0:5 Ys=c(2,2,2,3,3,3) result <- mapply(function(x,y) sum(x,y),Xs,Ys) result ##
[1] 2 3 4 6 7 8
ベクトル化された関数 汎用性が高い一部の関数で内部的に組み込まれた関数 ※ベクトル化された関数は一部なので、遅かれ早かれapply()ファミリーの適用が必要になる。 result=Xs+Ys # mapply()とsum()を適用しなくても、すでにベクトル化された“+”を使用できる。 result ## [1] 2 3
4 6 7 8 colSums(mat) # 列の和 ## [1] 9 3 6 2 3 3 rowSums(mat) # 行の和 ## [1] 12 3 5 6
参考:Rオンライン学習サイト DataCamp: Learn R, Python & Data Science Online Dataquest:
Learn R, Python and SQL for Data Science
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![データフレームの要素の抽出 列番号または名前で特定の列を抽出したり、行番号で特定の行を抽出したりできる。 mydata[,2:4] # 列番号2,3,4の列を抽出 mydata[,c("chr","start")] # 列名がchrとstartの列を抽出 mydata$start #](https://files.speakerdeck.com/presentations/b6834826a4c6415c85d42f86e1a2a51f/slide_14.jpg){kind=link}
{kind=link}
![リストの要素の抽出 位置または名前を二重括弧で指定して抽出できる。 # 位置(3番目)を指定してオブジェクトを抽出 w[[3]] ## [,1] [,2] ## [1,]](https://files.speakerdeck.com/presentations/b6834826a4c6415c85d42f86e1a2a51f/slide_16.jpg){kind=link}
![ファクター カテゴリーデータを格納するためのデータ構造 ※data.frame()でデータフレームを作成した場合に、文字列はデフォルトでファクターとして格納される。 features=c("promoter","exon","intron") f.feat=factor(features) # factor()関数でf.featにカテゴリーデータを格納 f.feat ## [1]](https://files.speakerdeck.com/presentations/b6834826a4c6415c85d42f86e1a2a51f/slide_17.jpg){kind=link}
![データ型 Rの4つのデータ型(numeric、logical、character、integer) # numericベクトルxを作成 x <- c(1,3,2,10,5) ## [1] 1](https://files.speakerdeck.com/presentations/b6834826a4c6415c85d42f86e1a2a51f/slide_18.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
![ベクトル化された関数 汎用性が高い一部の関数で内部的に組み込まれた関数 ※ベクトル化された関数は一部なので、遅かれ早かれapply()ファミリーの適用が必要になる。 result=Xs+Ys # mapply()とsum()を適用しなくても、すでにベクトル化された“+”を使用できる。 result ## [1] 2 3](https://files.speakerdeck.com/presentations/b6834826a4c6415c85d42f86e1a2a51f/slide_42.jpg){kind=link}
{kind=link}