to: • Assess HGAP assembly results of bacterial genomes Scientists, Research Associates, Bioinformaticians: • Interested in finishing and closing bacterial genomes • Familiarity with UNIX commands
of contigs – N50: Equal to the size of the contig found if you sort contigs by size and walk to the contig that represents 50% of the total sequence − N50 = 10 bp − Mean contig length = 3 bp – Max contig size • Limitation of these metrics: – They do not capture information about assembly accuracy! − Large scale mis-assemblies − Base level errors – There might be more than one chromosome (plasmid, phage, etc.) – Contaminants may contribute to a contig number (such as a cloning vector) 4 10 4 1 1 1 1
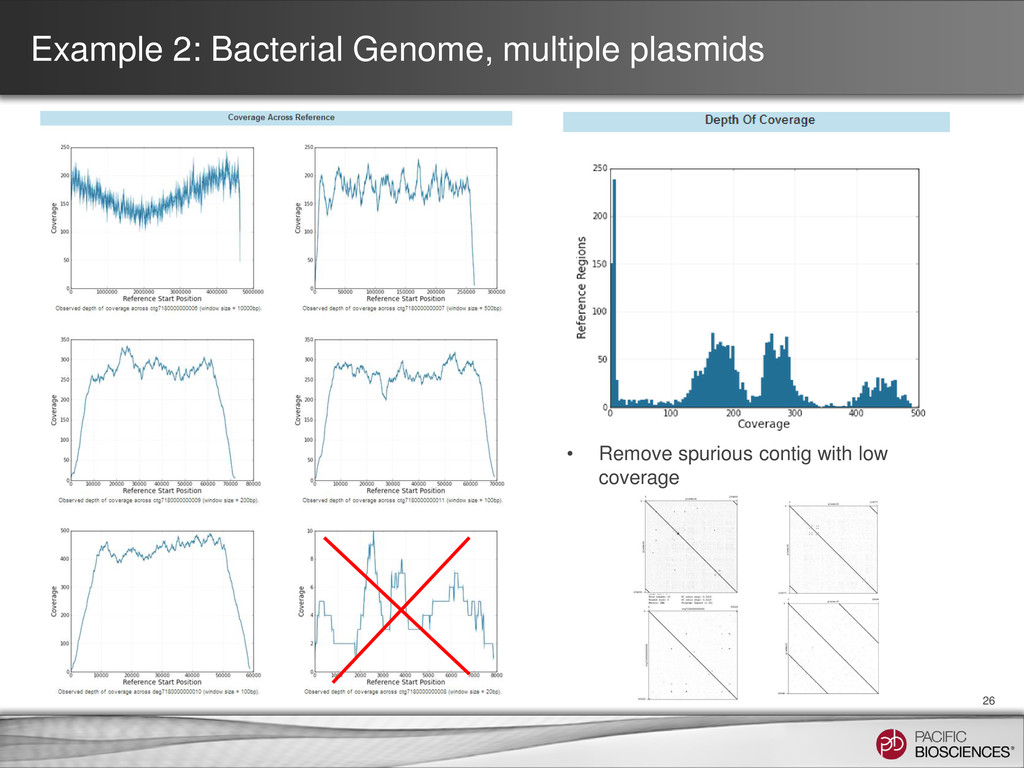
Undulation in coverage in chromosome is biological (more DNA close to ori when cells are harvested in log phase) • Different levels of coverage between chromosome and one of the plasmids, leading to distinct coverage peaks in histogram ori
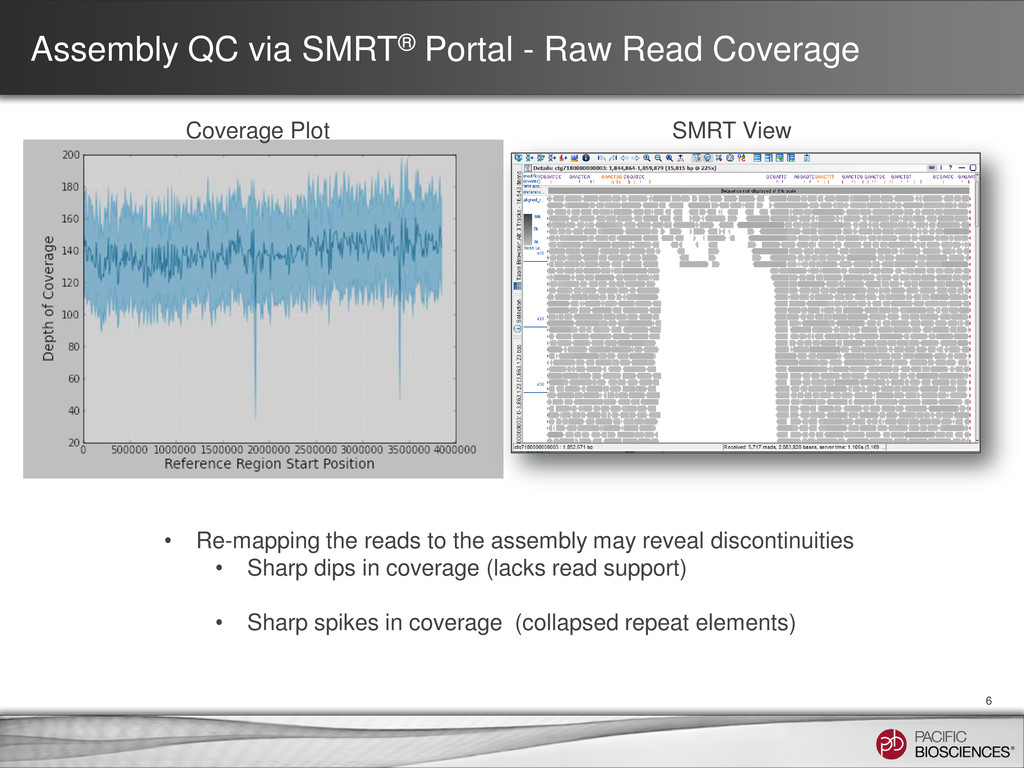
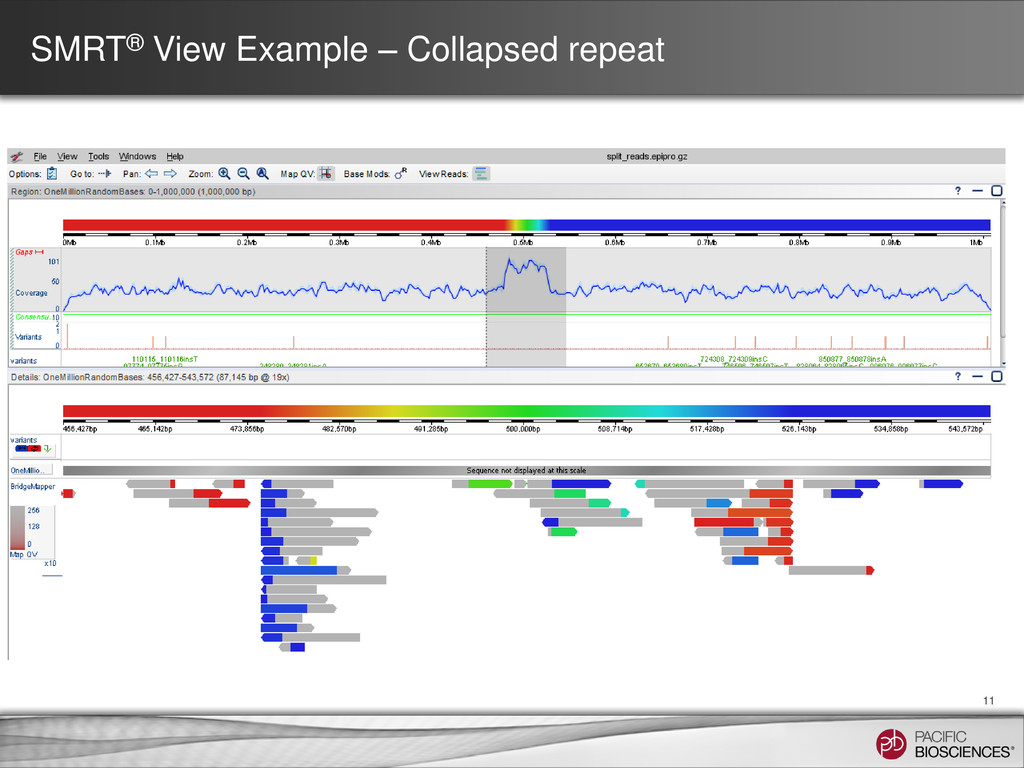
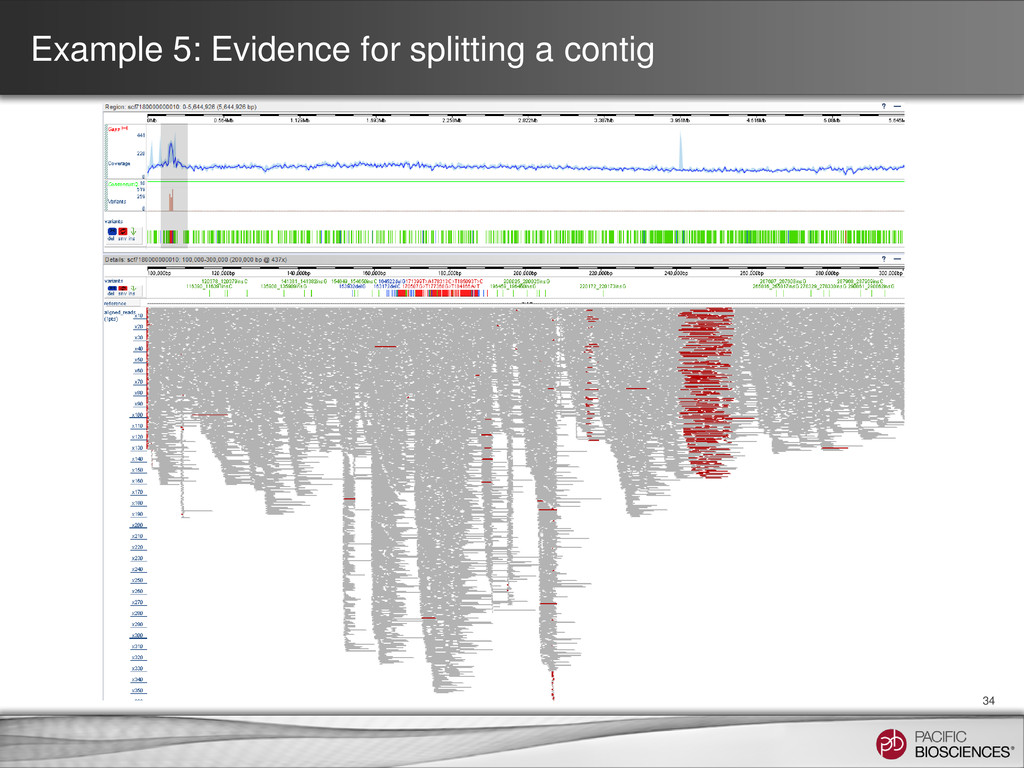
Coverage Plot SMRT View • Re-mapping the reads to the assembly may reveal discontinuities • Sharp dips in coverage (lacks read support) • Sharp spikes in coverage (collapsed repeat elements)
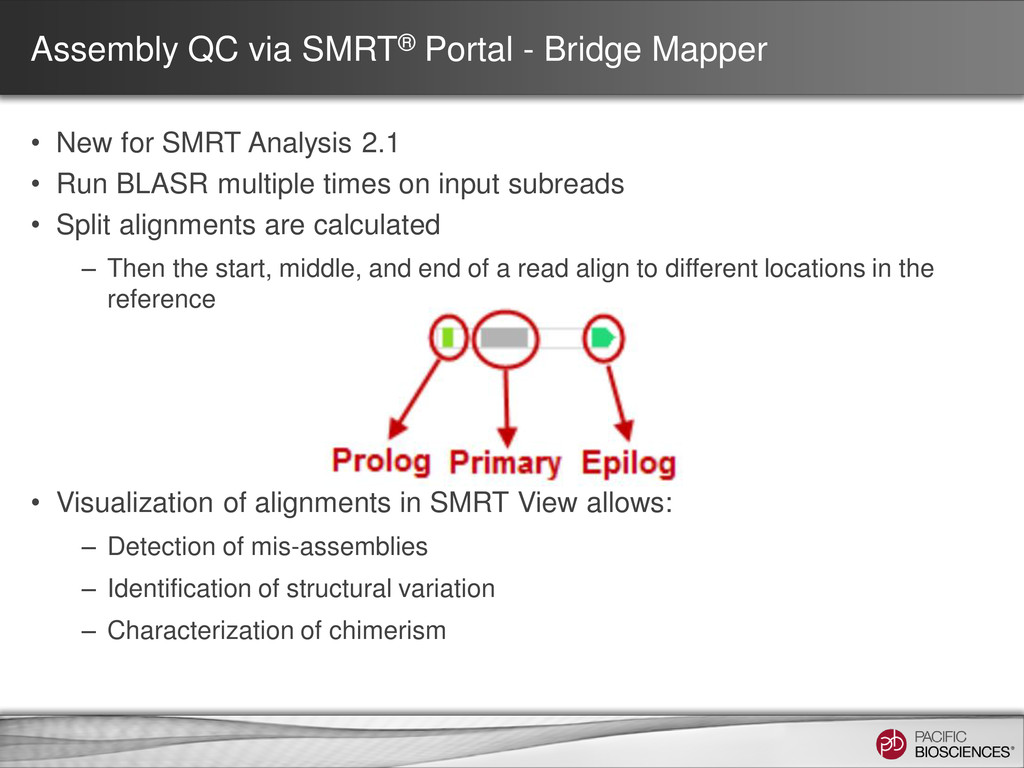
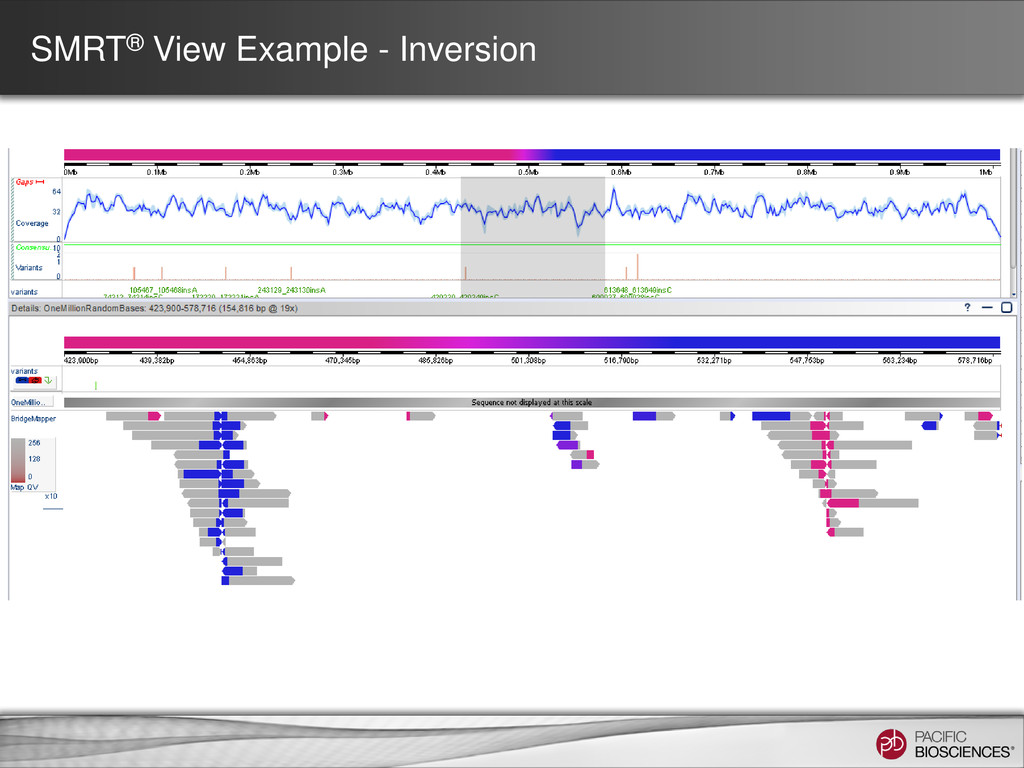
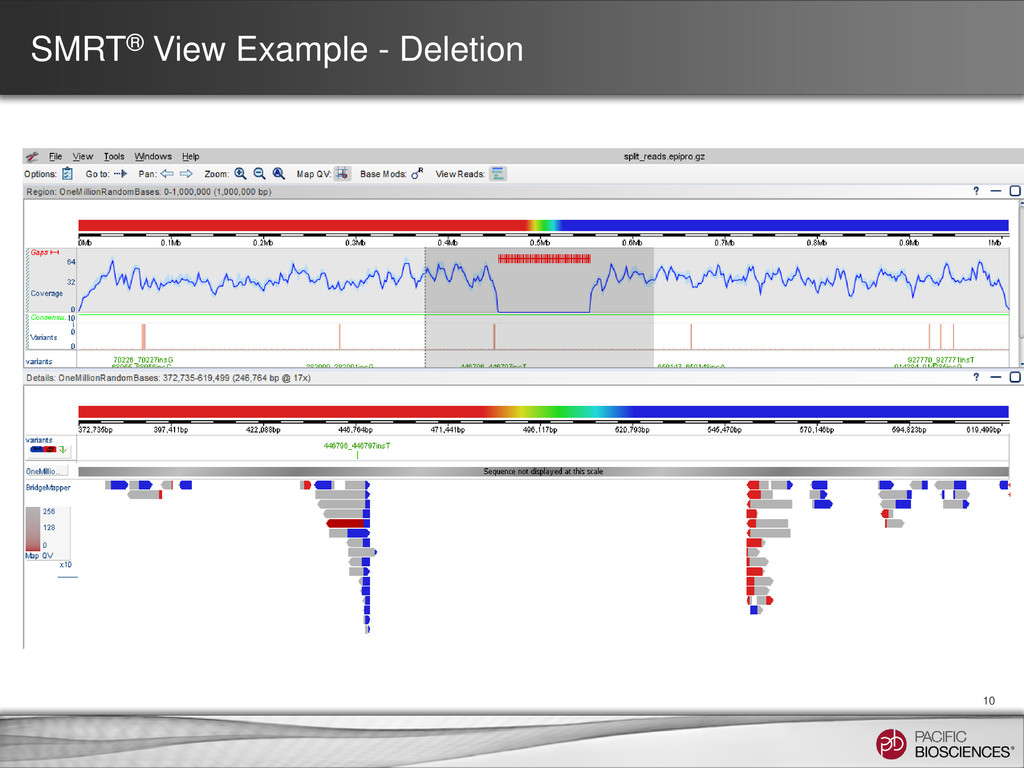
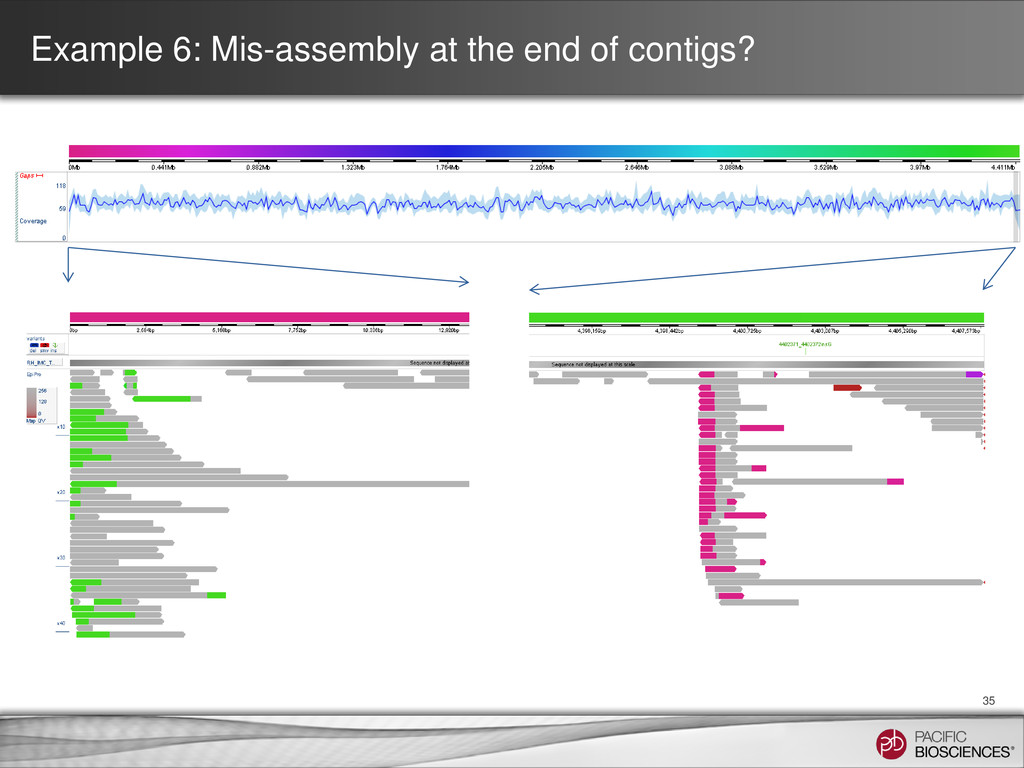
for SMRT Analysis 2.1 • Run BLASR multiple times on input subreads • Split alignments are calculated – Then the start, middle, and end of a read align to different locations in the reference • Visualization of alignments in SMRT View allows: – Detection of mis-assemblies – Identification of structural variation – Characterization of chimerism
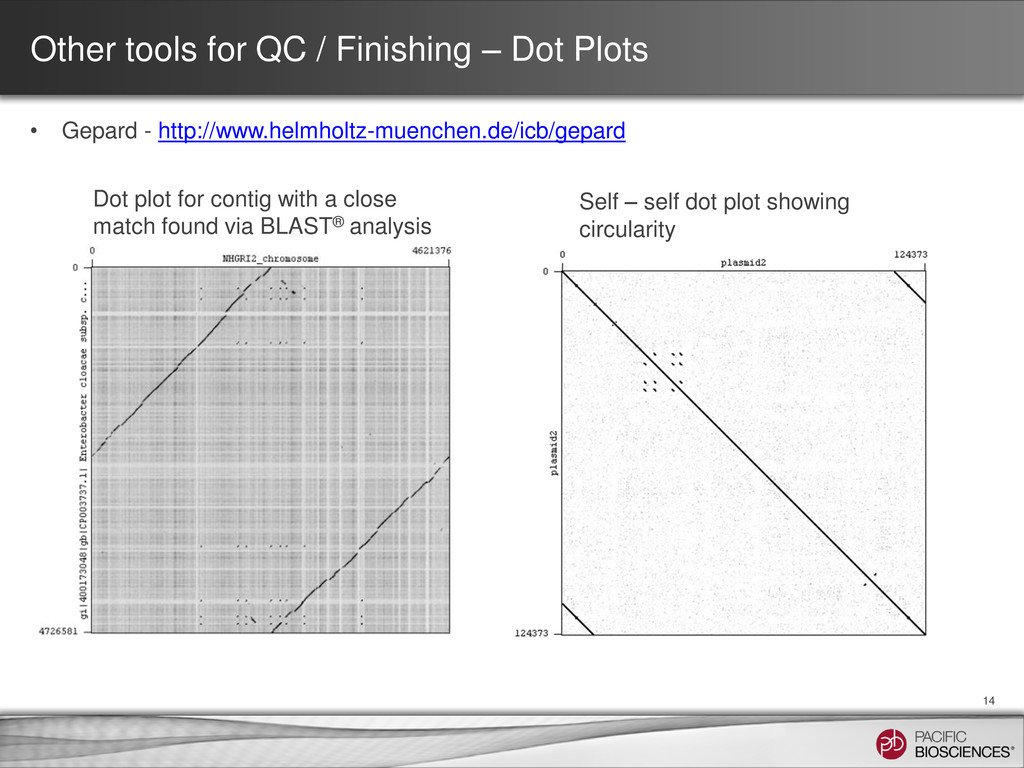
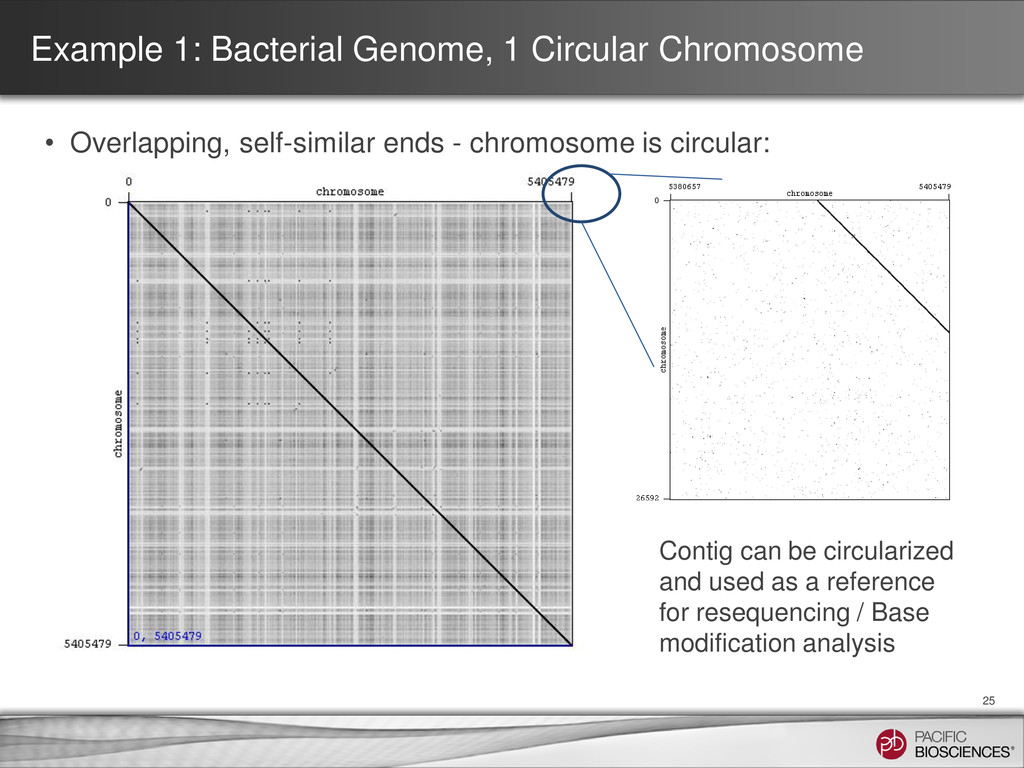
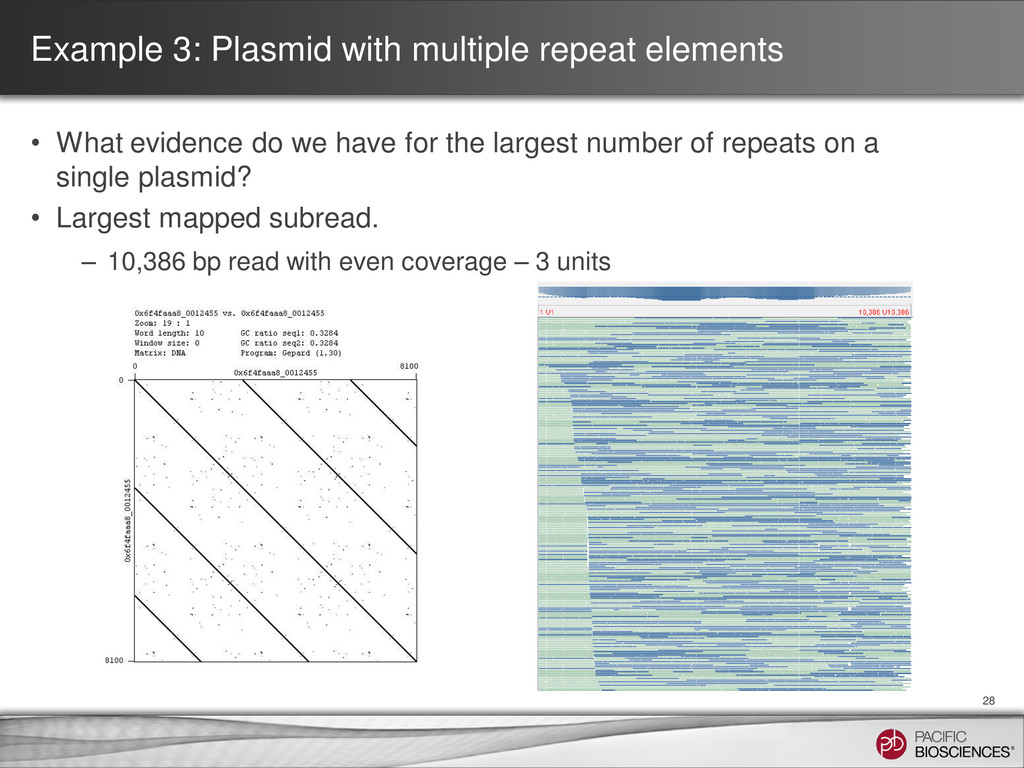
Dot plot for contig with a close match found via BLAST® analysis • Gepard - http://www.helmholtz-muenchen.de/icb/gepard Self – self dot plot showing circularity
Split Contig Overlap Consensus Manually introduce a break, “>”, in the fasta sequence Minimus2 can be used as a simple overlapper. Minimus2 - http://sourceforge.net/apps/mediawiki/amos/i ndex.php?title=Minimus2
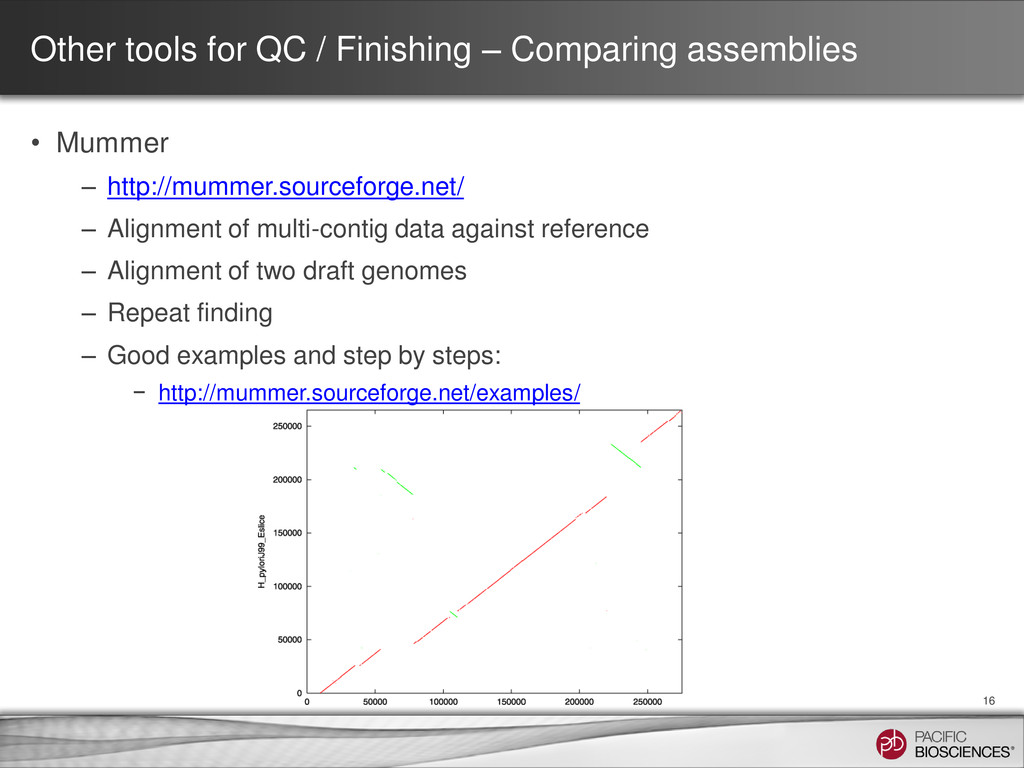
Mummer – http://mummer.sourceforge.net/ – Alignment of multi-contig data against reference – Alignment of two draft genomes – Repeat finding – Good examples and step by steps: − http://mummer.sourceforge.net/examples/ 16
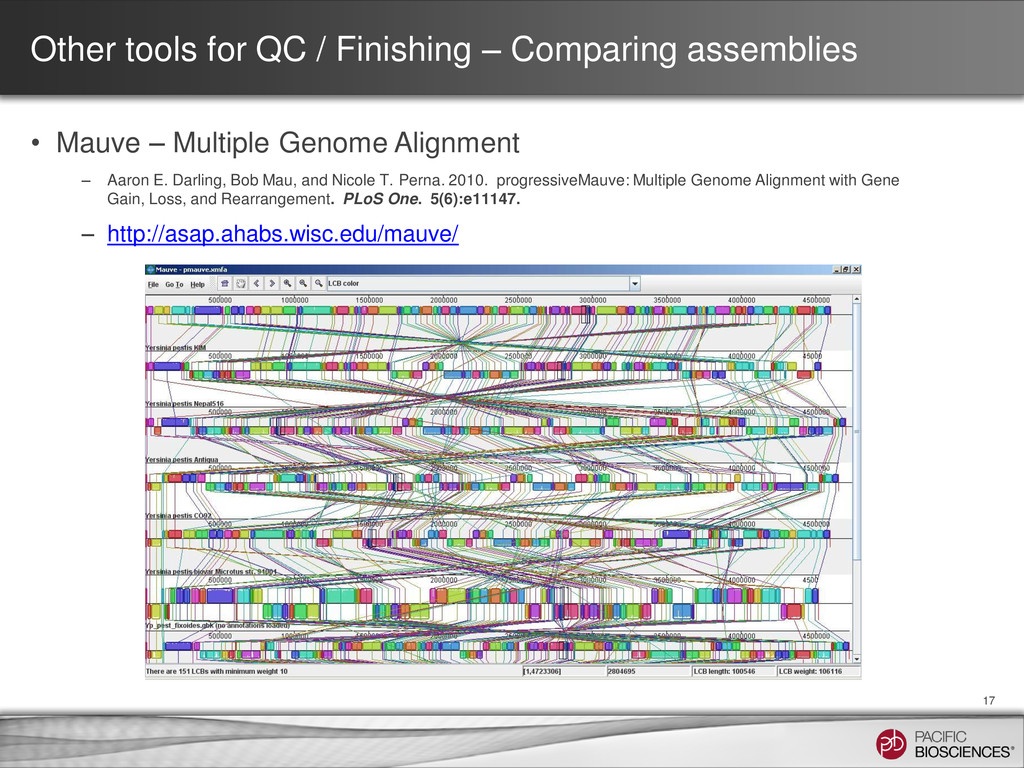
Mauve – Multiple Genome Alignment – Aaron E. Darling, Bob Mau, and Nicole T. Perna. 2010. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss, and Rearrangement. PLoS One. 5(6):e11147. – http://asap.ahabs.wisc.edu/mauve/ 17
in the chromosome • Regions 2, 6 and 8 each have a single adenine-specific methyltransferase using PHAST (http://phast.wishartlab.com) Region2 Region6 Region8
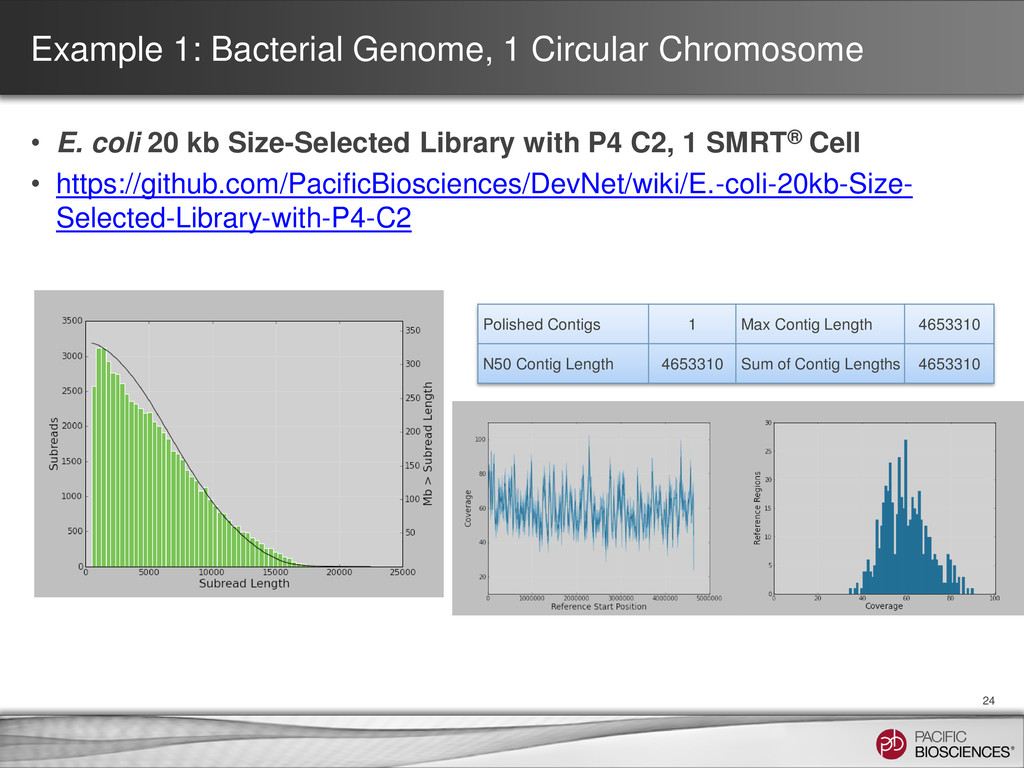
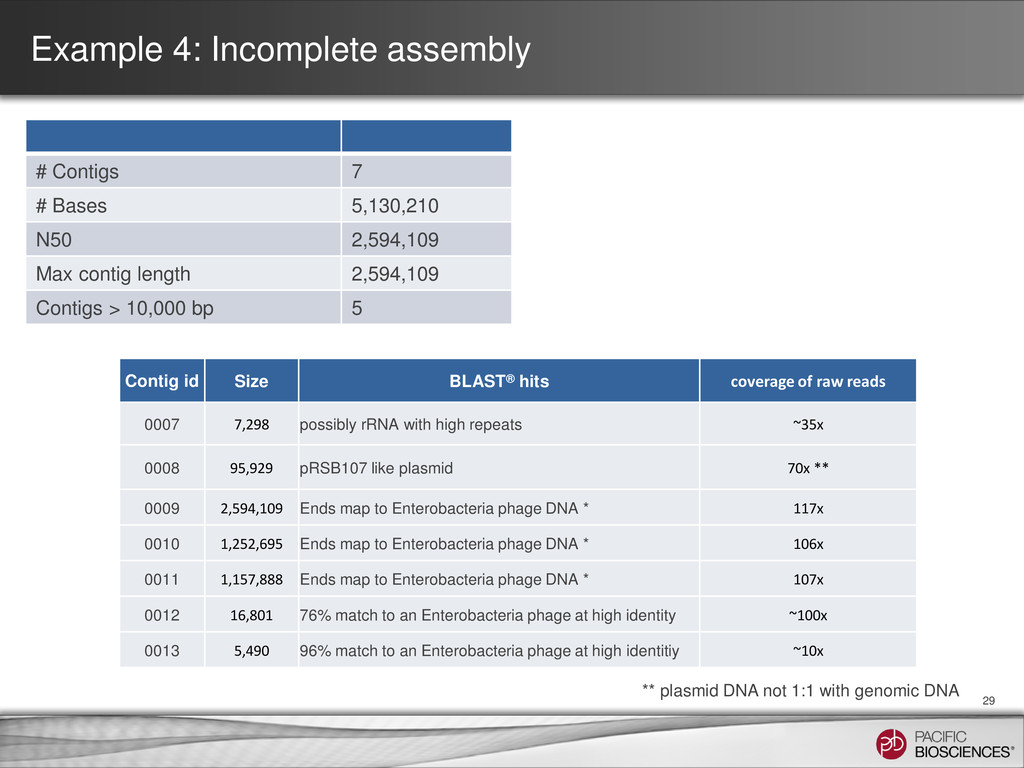
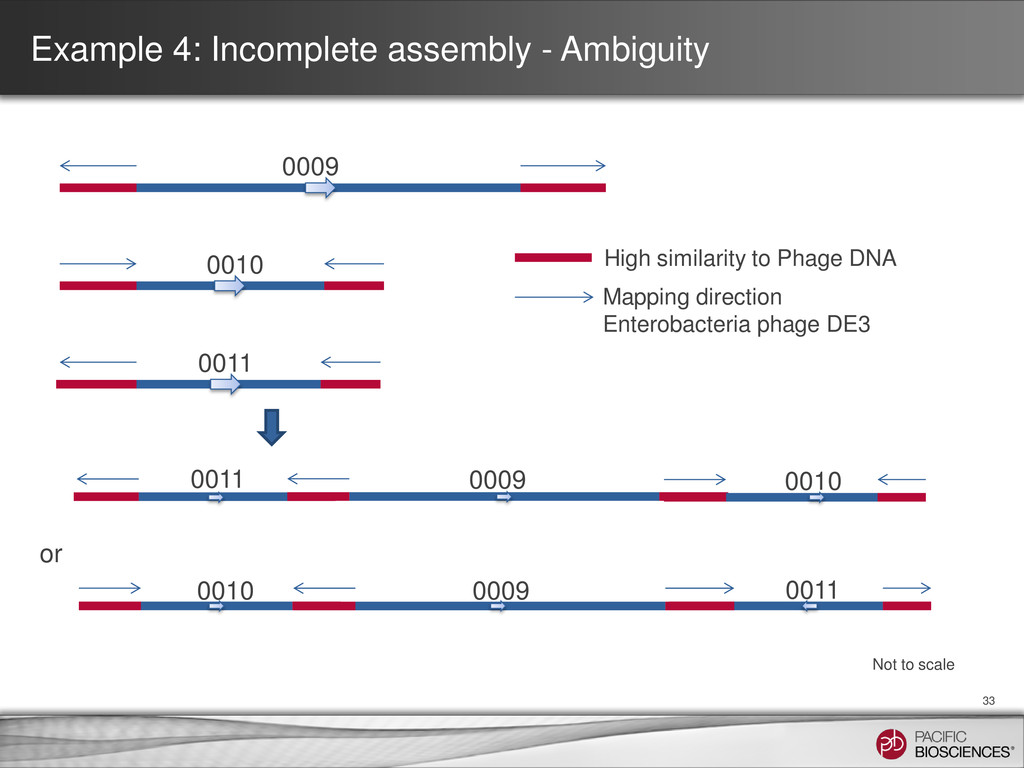
5,130,210 N50 2,594,109 Max contig length 2,594,109 Contigs > 10,000 bp 5 Contig id Size BLAST® hits coverage of raw reads 0007 7,298 possibly rRNA with high repeats ~35x 0008 95,929 pRSB107 like plasmid 70x ** 0009 2,594,109 Ends map to Enterobacteria phage DNA * 117x 0010 1,252,695 Ends map to Enterobacteria phage DNA * 106x 0011 1,157,888 Ends map to Enterobacteria phage DNA * 107x 0012 16,801 76% match to an Enterobacteria phage at high identity ~100x 0013 5,490 96% match to an Enterobacteria phage at high identitiy ~10x ** plasmid DNA not 1:1 with genomic DNA
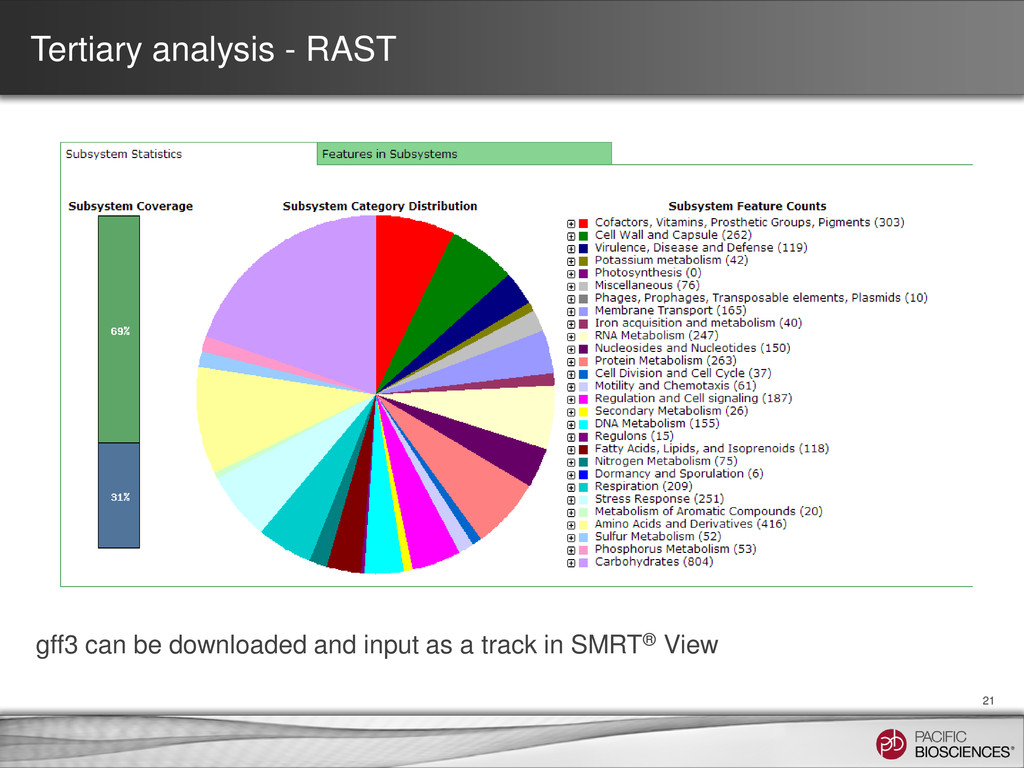
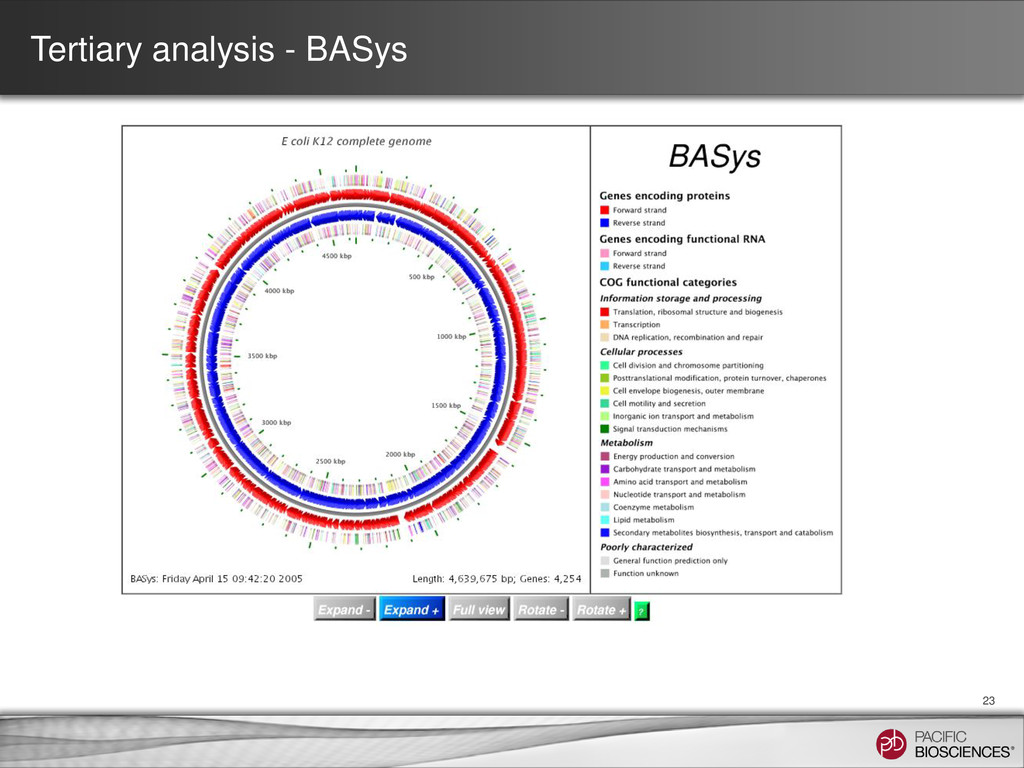
contig numbers are low • SMRT® Portal can be used as a first pass – Coverage plot – SMRT View − Raw read coverage − Bridge Mapping • Third party tools can be used for QC / finishing – Dot plots – Aligning to know sequences – Circularization • Tertiary analysis for bacterial genomes can be done in an automated fashion and results visualized in SMRT View – RAST, PHAST, BASys 37
are trademarks of Pacific Biosciences in the United States and/or other countries. All other trademarks are the sole property of their respective owners.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}