(HHMI / The Salk Institute ) -Alicia Clum, Kerrie Barry, Alex Copeland (Joint Genome Institute) -Maria Nattestad, Fritz Sedlazeck, Michael Schatz (CSHL) - Open source toolsets -Daligner, Gene Myers -Blasr, Mark Chaisson -Python, NetworkX for rapid algorithm protyping -Gephi, Graphviz for graph visualization
- N50 ~ 10 Mb to 30 Mb, depending on DNA sample and sequencing quality - Longest contig that we ever assembled ~ 109 Mb http://www.pacb.com/blog/toward-platinum-genomes-pacbio-releases-a-new-higher-quality-chm1-assembly-to-ncbi/ Google search, “pacbio chm1 assembly blog”
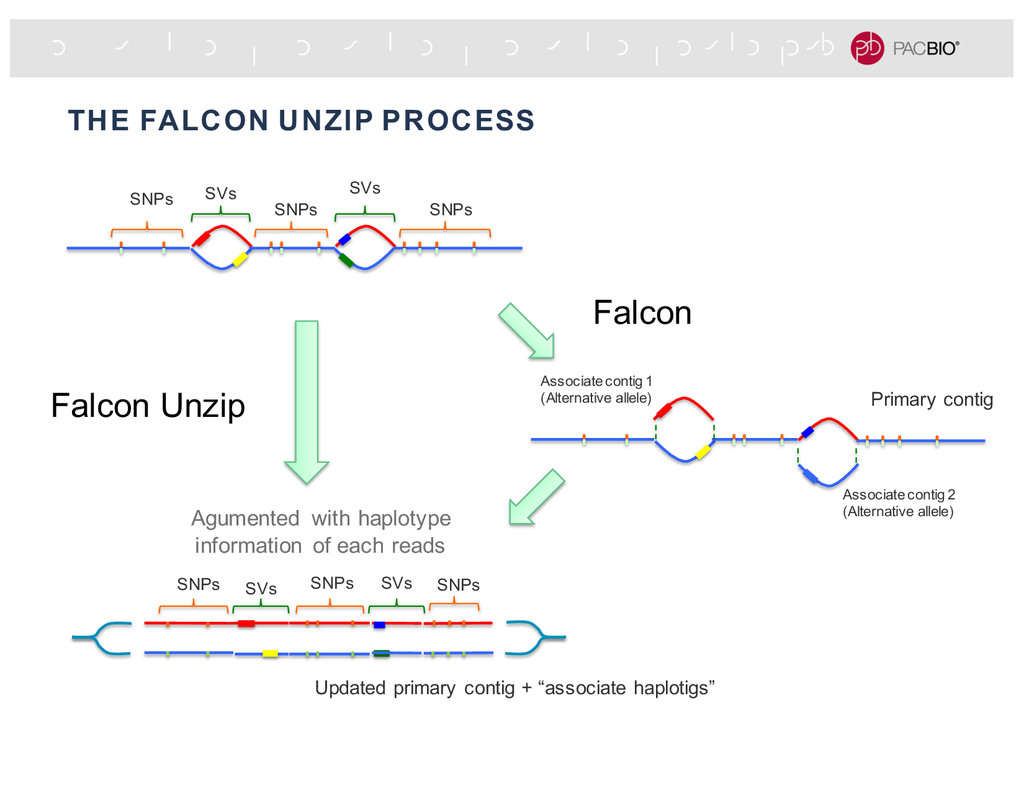
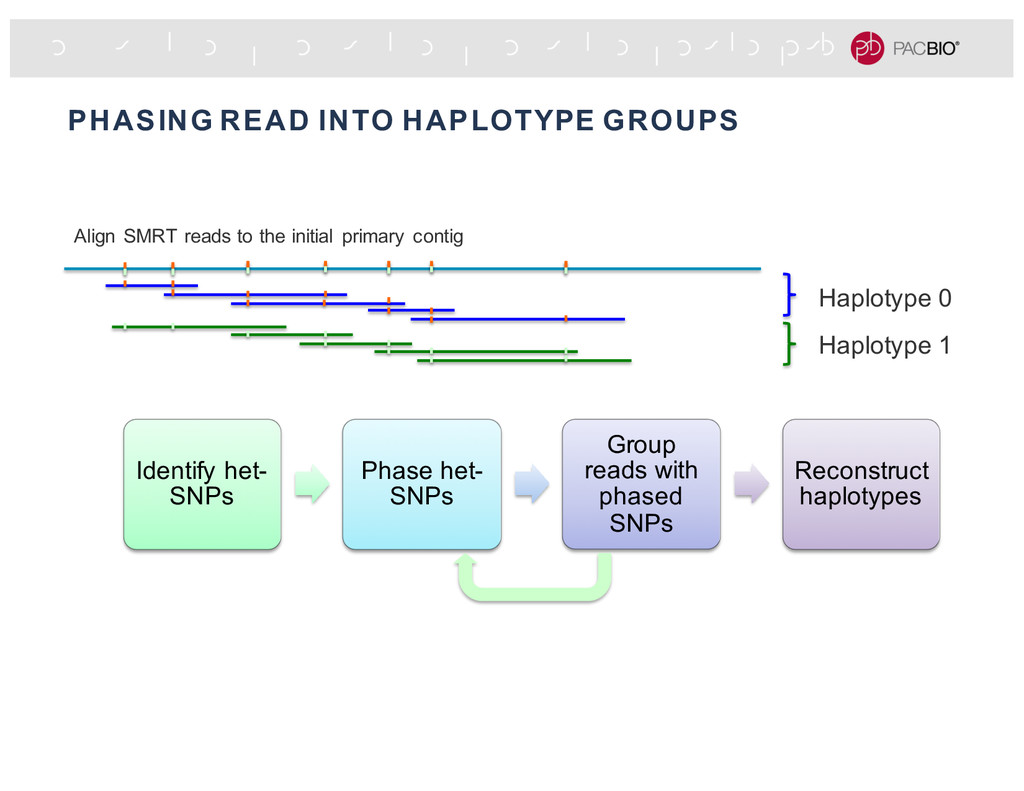
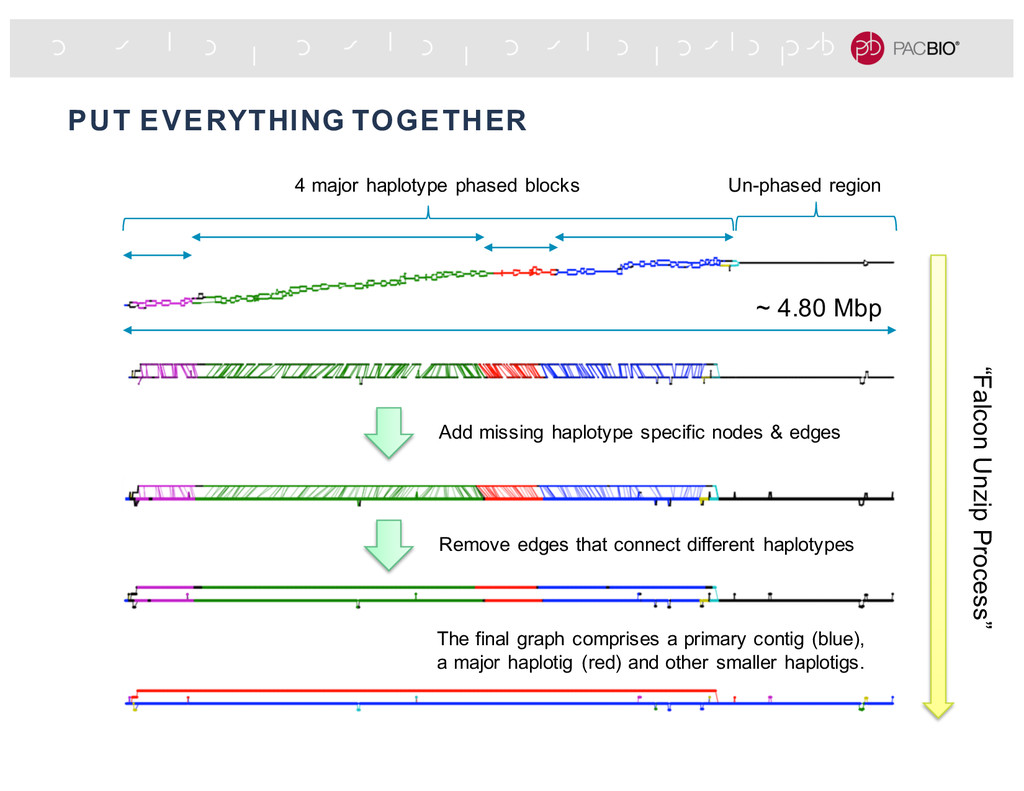
missing haplotype specific nodes & edges Remove edges that connect different haplotypes The final graph comprises a primary contig (blue), a major haplotig (red) and other smaller haplotigs. 4 major haplotype phased blocks Un-phased region
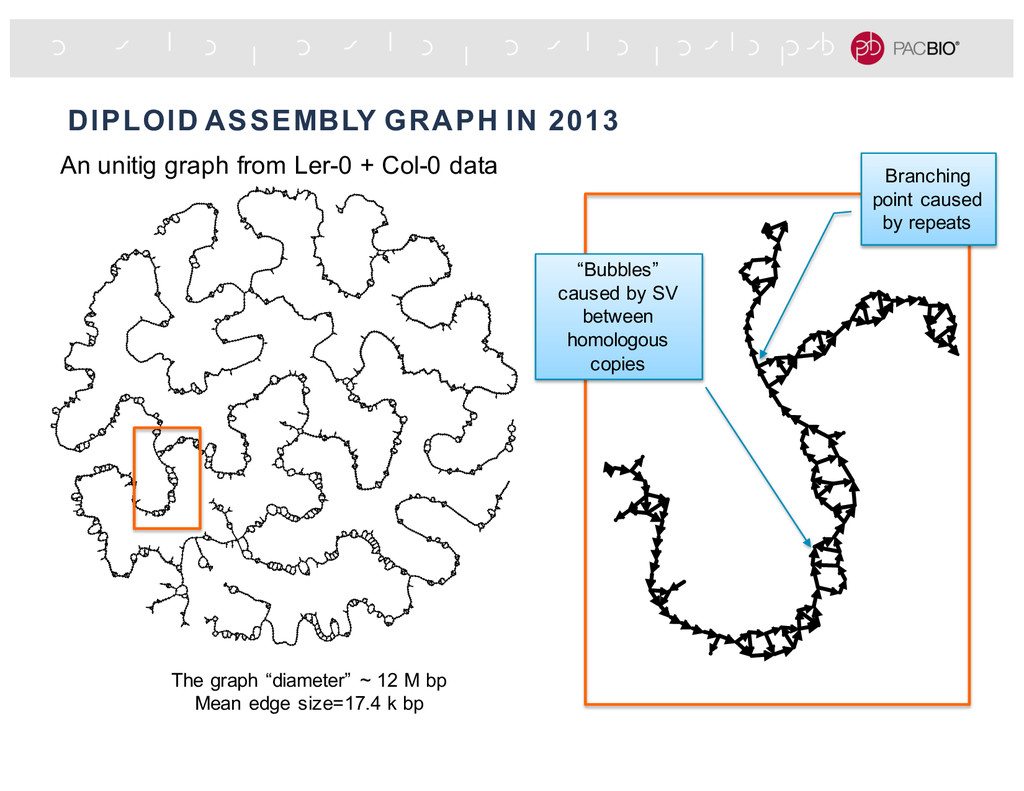
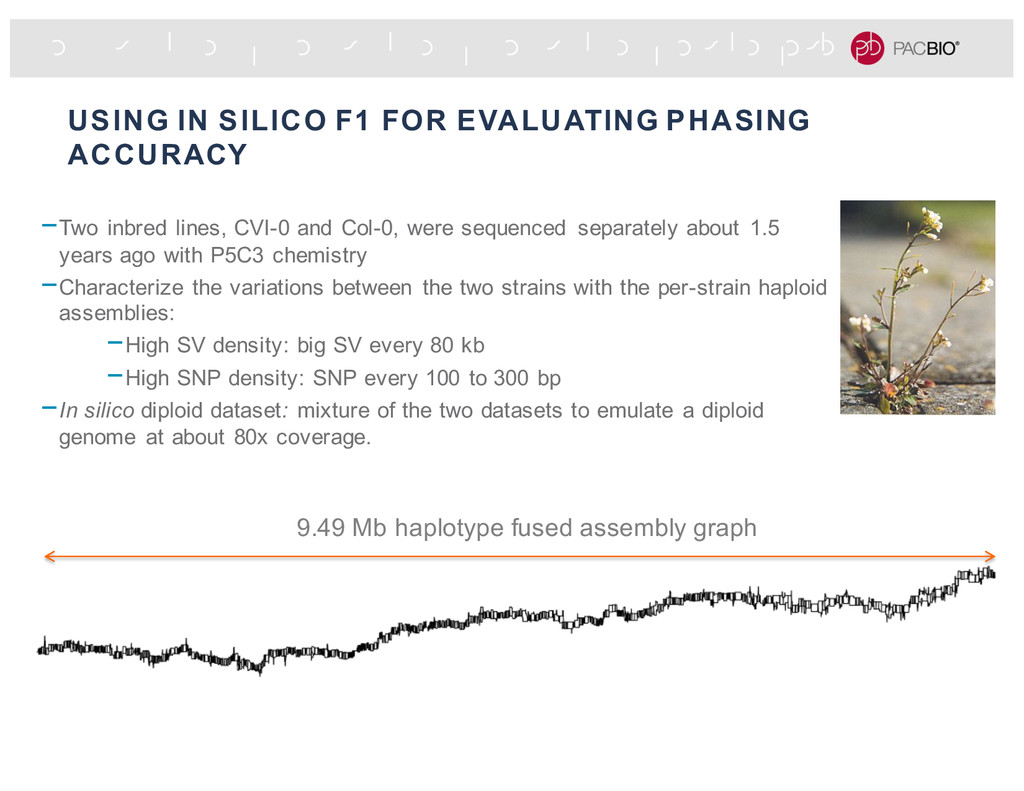
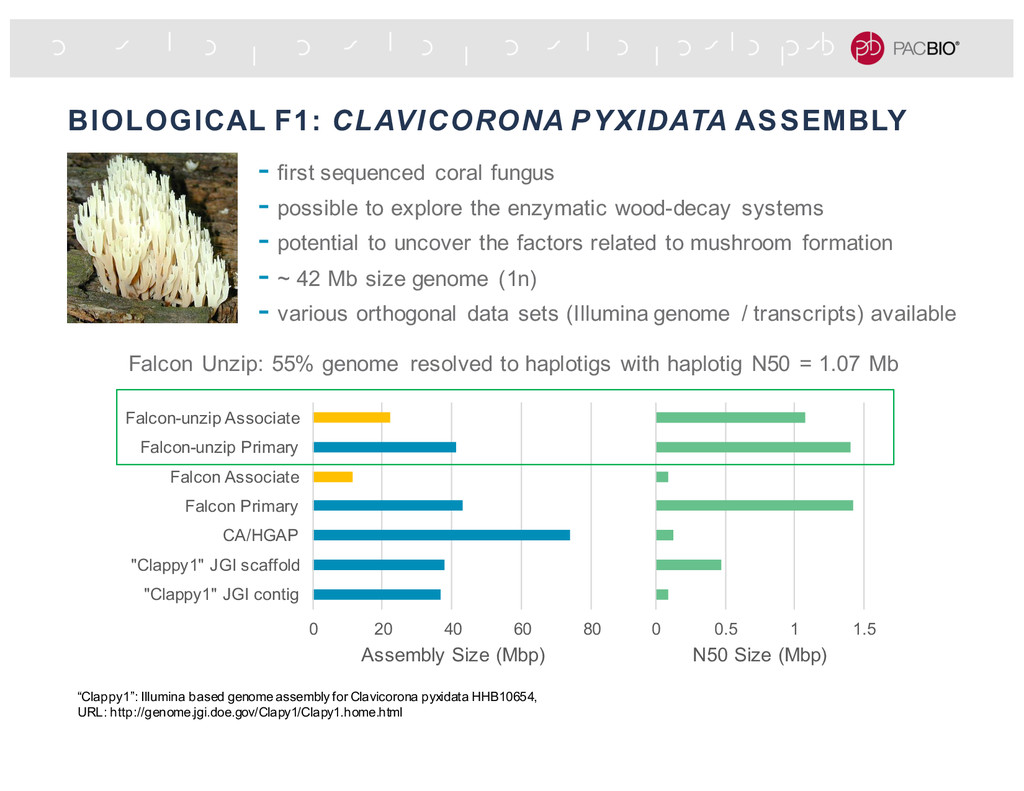
lines, CVI-0 and Col-0, were sequenced separately about 1.5 years ago with P5C3 chemistry -Characterize the variations between the two strains with the per-strain haploid assemblies: -High SV density: big SV every 80 kb -High SNP density: SNP every 100 to 300 bp -In silico diploid dataset: mixture of the two datasets to emulate a diploid genome at about 80x coverage. 9.49 Mb haplotype fused assembly graph
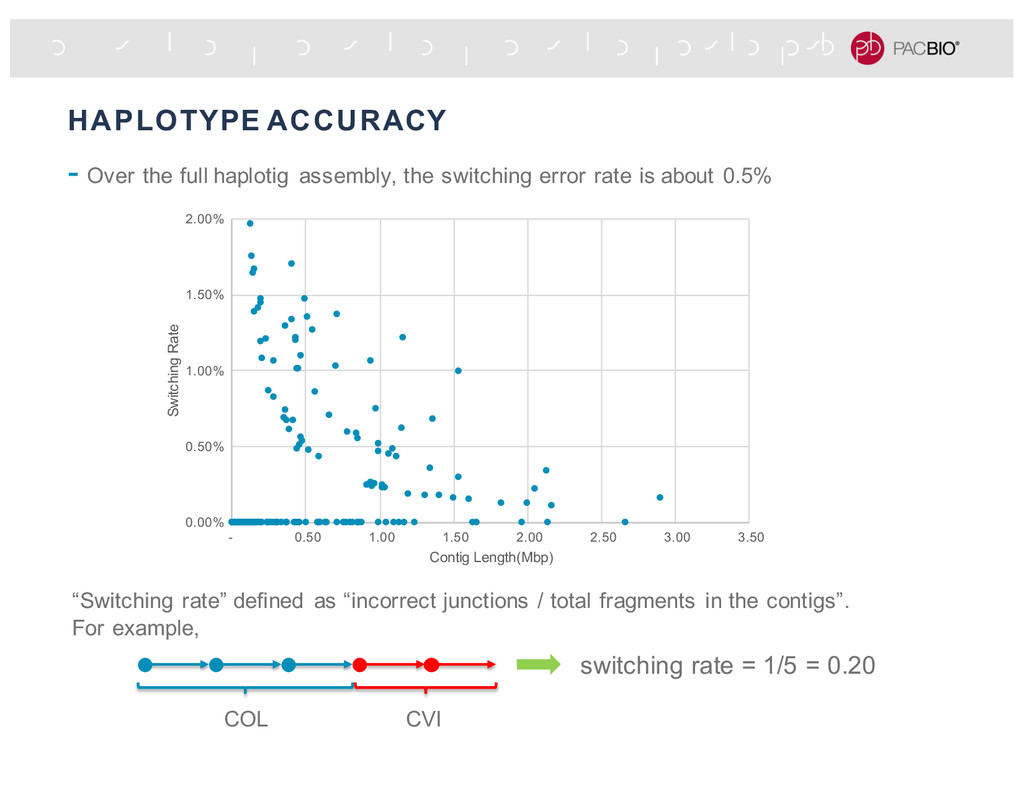
1.50 2.00 2.50 3.00 3.50 Switching Rate Contig Length(Mbp) - Over the full haplotig assembly, the switching error rate is about 0.5% “Switching rate” defined as “incorrect junctions / total fragments in the contigs”. For example, switching rate = 1/5 = 0.20 COL CVI
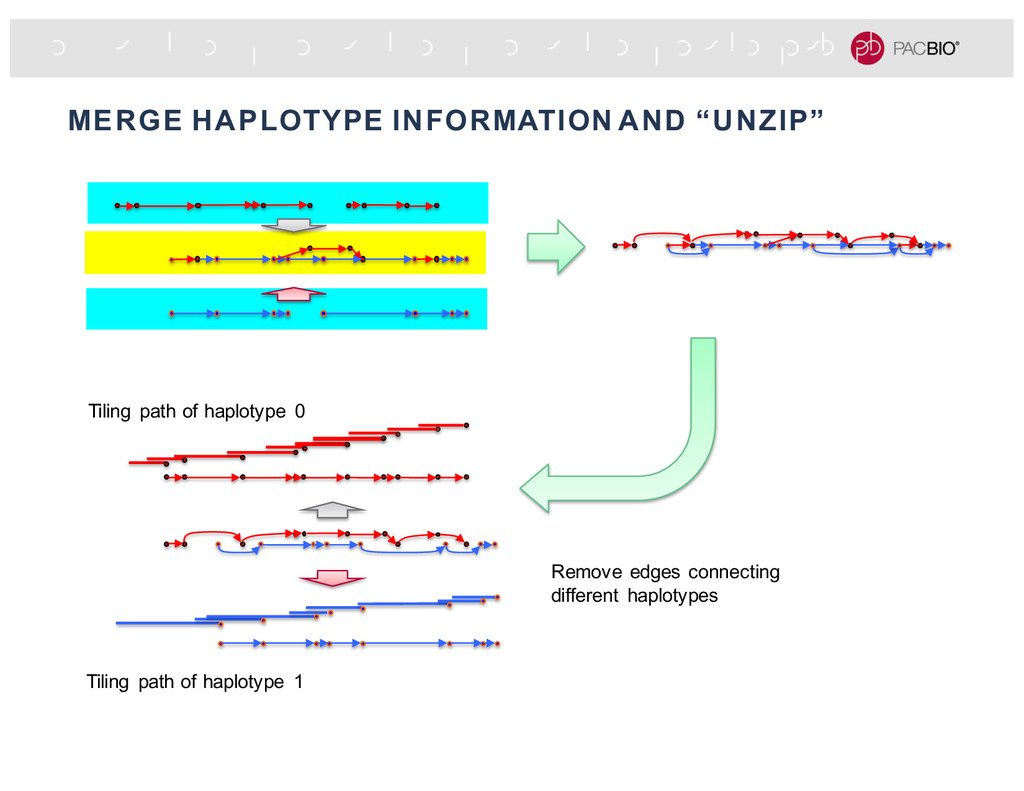
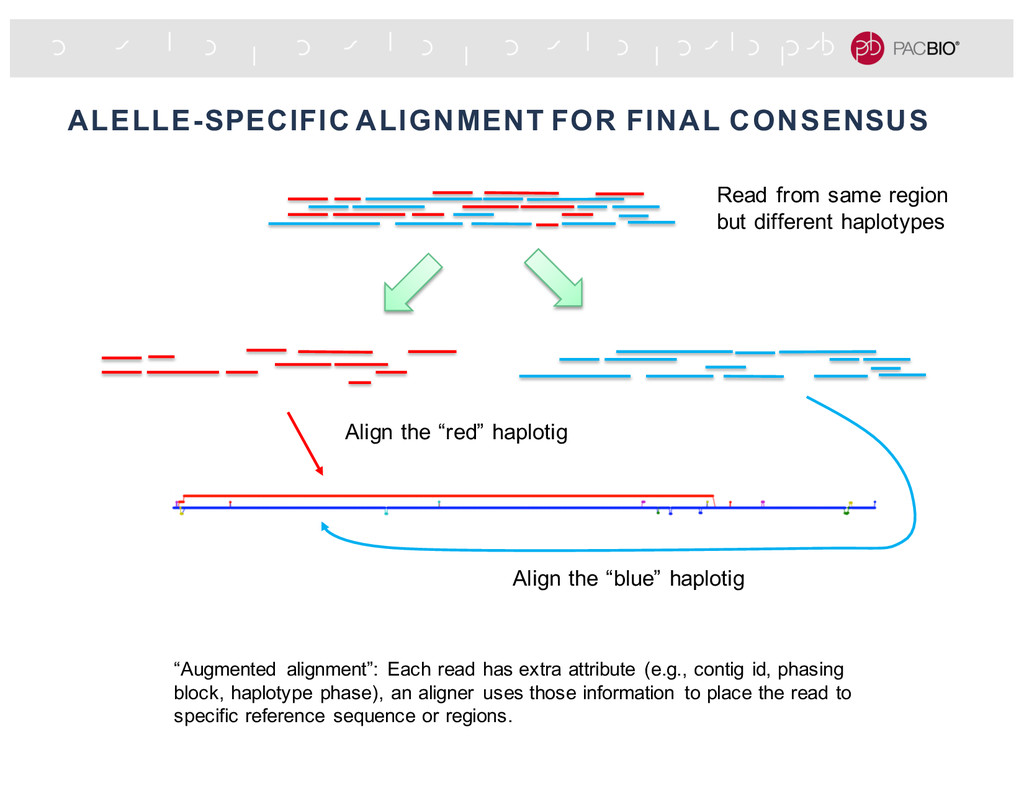
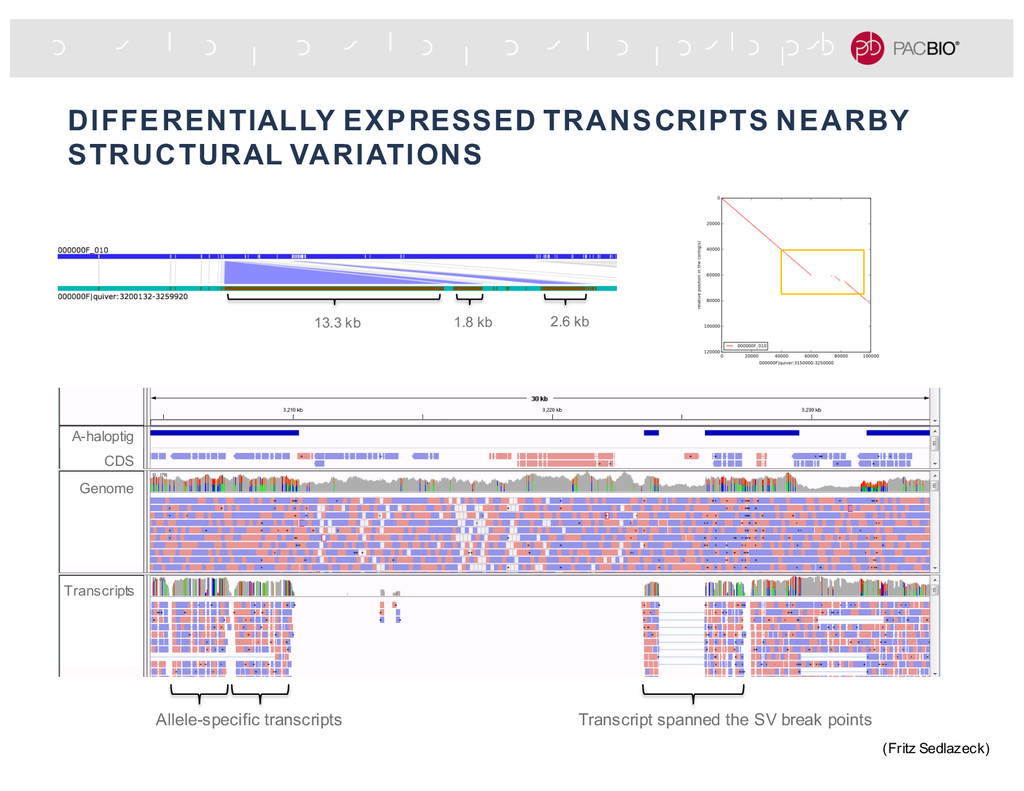
extra attribute (e.g., contig id, phasing block, haplotype phase), an aligner uses those information to place the read to specific reference sequence or regions. Align the “red” haplotig Align the “blue” haplotig Read from same region but different haplotypes
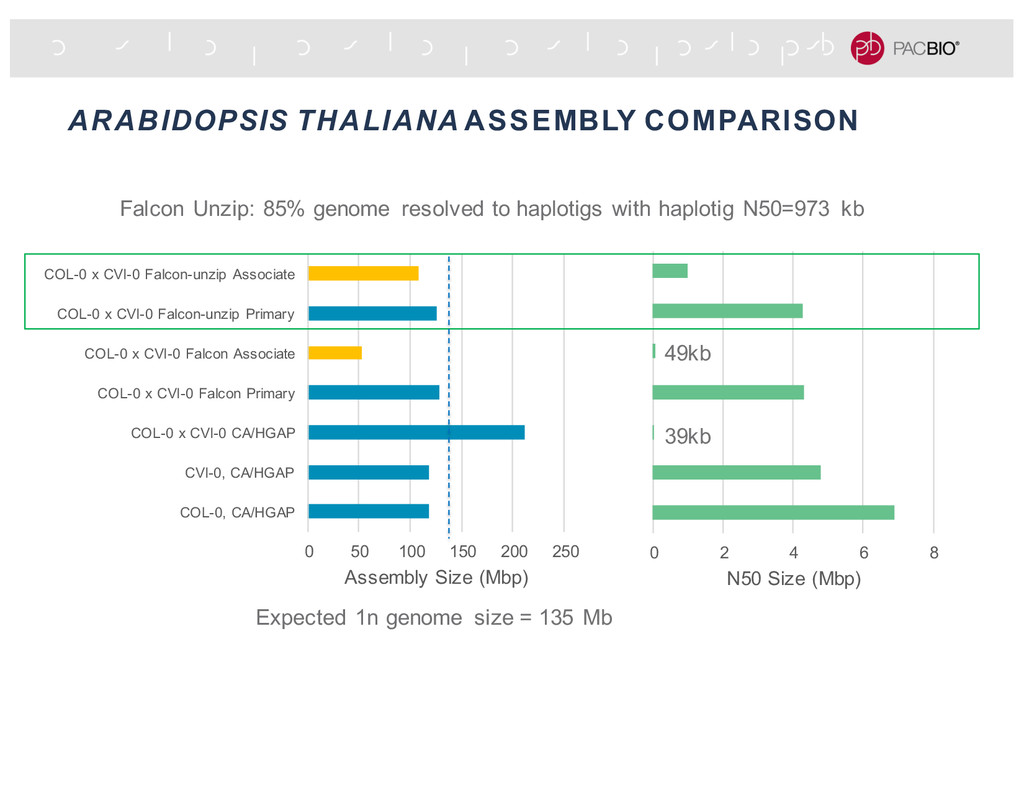
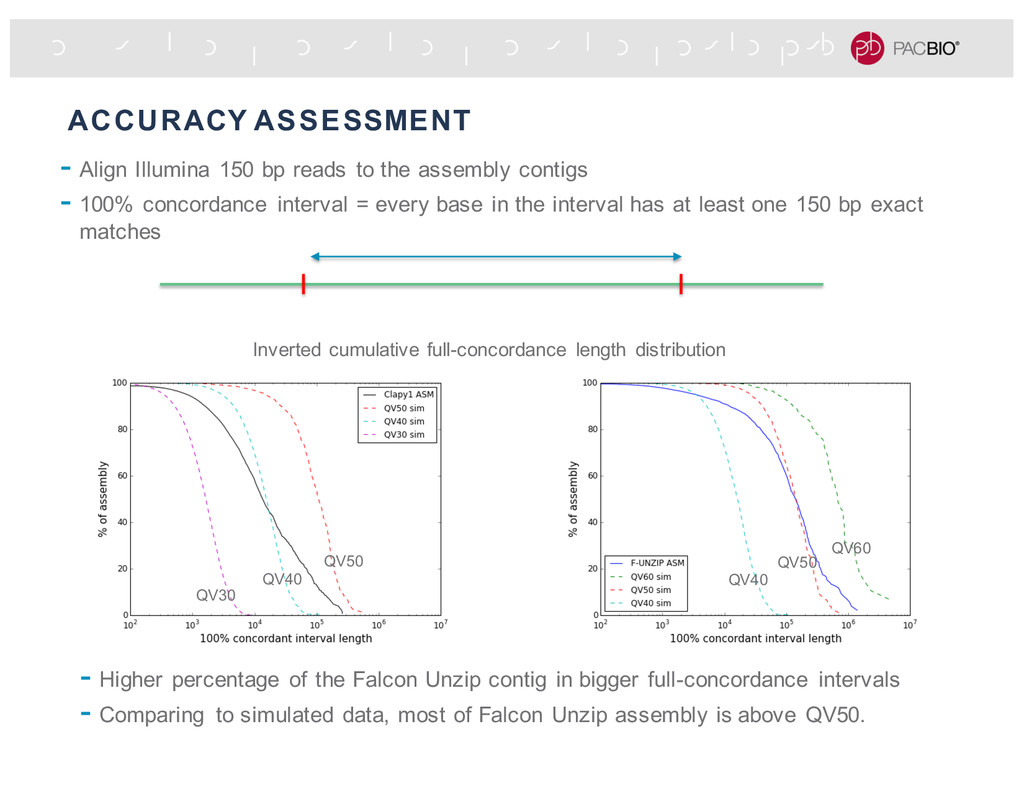
assembly contigs - 100% concordance interval = every base in the interval has at least one 150 bp exact matches - Higher percentage of the Falcon Unzip contig in bigger full-concordance intervals - Comparing to simulated data, most of Falcon Unzip assembly is above QV50. QV50 QV40 QV30 QV50 QV60 QV40 Inverted cumulative full-concordance length distribution
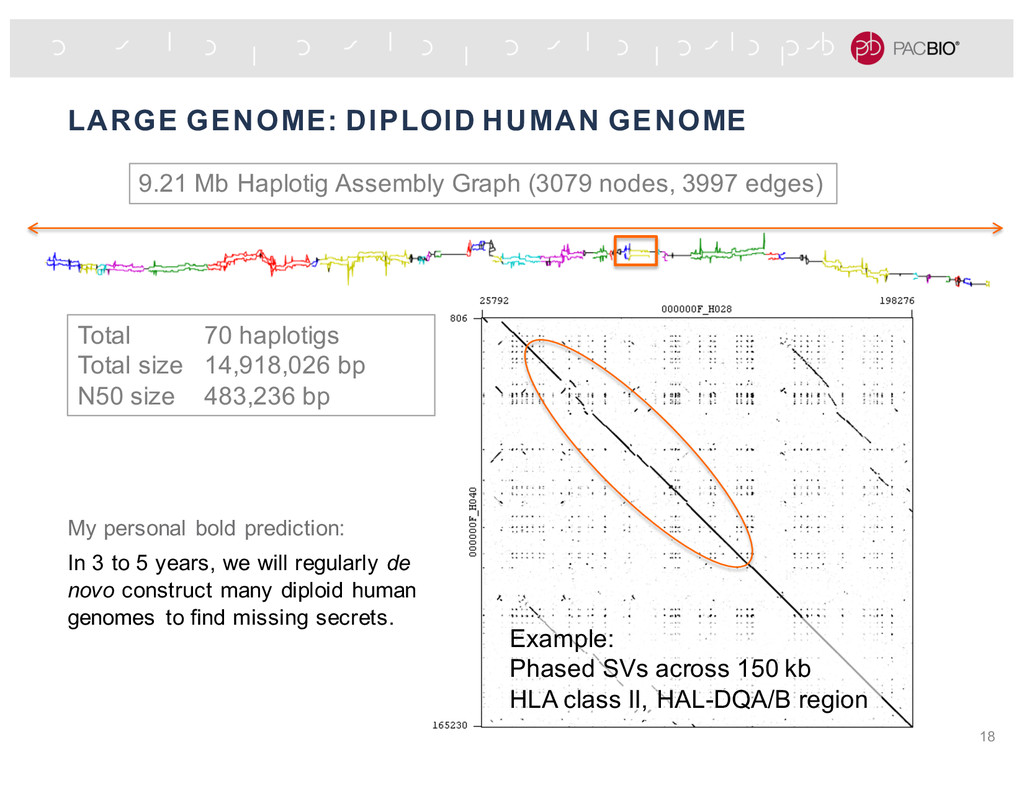
Graph (3079 nodes, 3997 edges) Total 70 haplotigs Total size 14,918,026 bp N50 size 483,236 bp Example: Phased SVs across 150 kb HLA class II, HAL-DQA/B region My personal bold prediction: In 3 to 5 years, we will regularly de novo construct many diploid human genomes to find missing secrets.
We need to keep developing evaluation frameworks to improve the performance - Large genomes are challenging but it is mostly an engineering problem now: -Haplotype phasing improvement, incorporate 3rd party phasing code -Develop a sequence aligner for “augmented alignment” for faster Quiver consensus process -Want to attack polyploid genome assembly problem? Let us help you!
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}