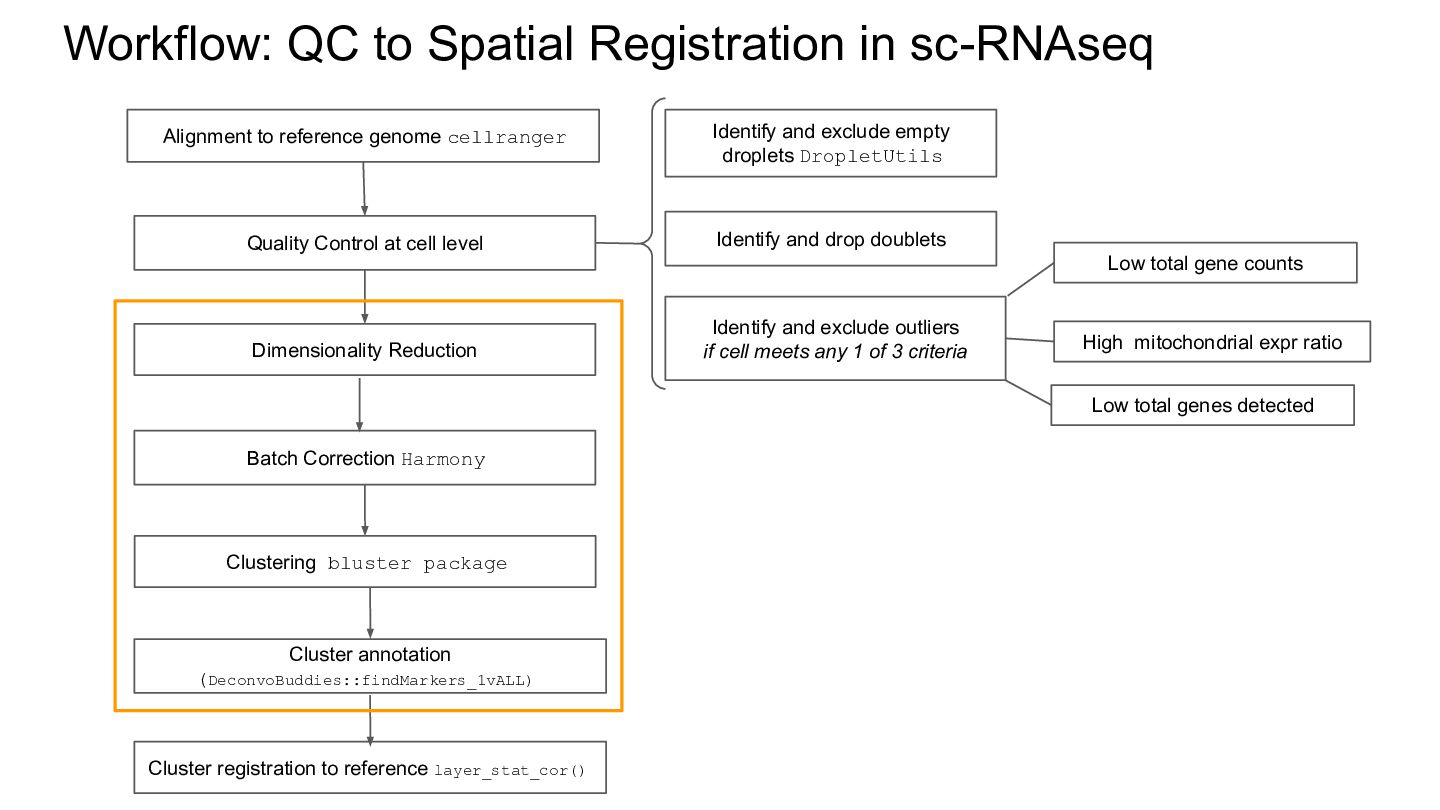
genome cellranger Quality Control at cell level Identify and exclude empty droplets DropletUtils High mitochondrial expr ratio Low total gene counts Identify and exclude outliers if cell meets any 1 of 3 criteria Low total genes detected Identify and drop doublets Cluster registration to reference layer_stat_cor() Cluster annotation (DeconvoBuddies::findMarkers_1vALL) Dimensionality Reduction Batch Correction Harmony Clustering bluster package
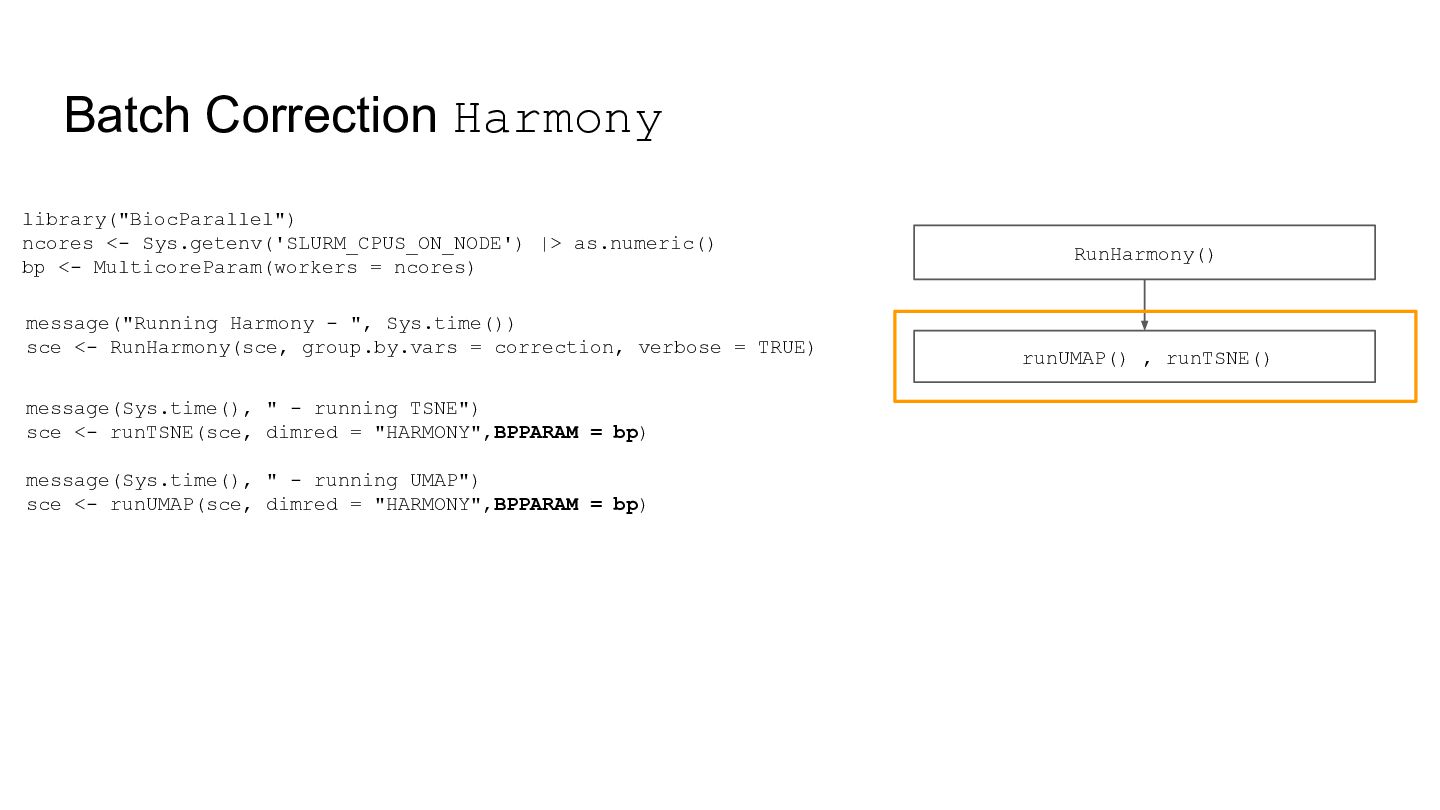
addition to the std values. Note: setting this to TRUE roughly doubles run time. library("BiocParallel") library("DeconvoBuddies") ncores <- Sys.getenv('SLURM_CPUS_ON_NODE') |> as.numeric() bp <- MulticoreParam(workers = ncores) annotation <- findMarkers_1vAll( sce, assay_name = "logcounts", cellType_col = "cellType", #input_k_resolution, BPPARAM = bp ) *raw_logFC: default FALSE Calculate 1 vs. All standard fold change for each gene x cell type
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}