2 Experimental-design guidelines by genome size Sample-preparation and sequencing recommendations Assembly options with PacBio data PacBio® benefits for finishing genomes Assembly recommendations Where to find additional information
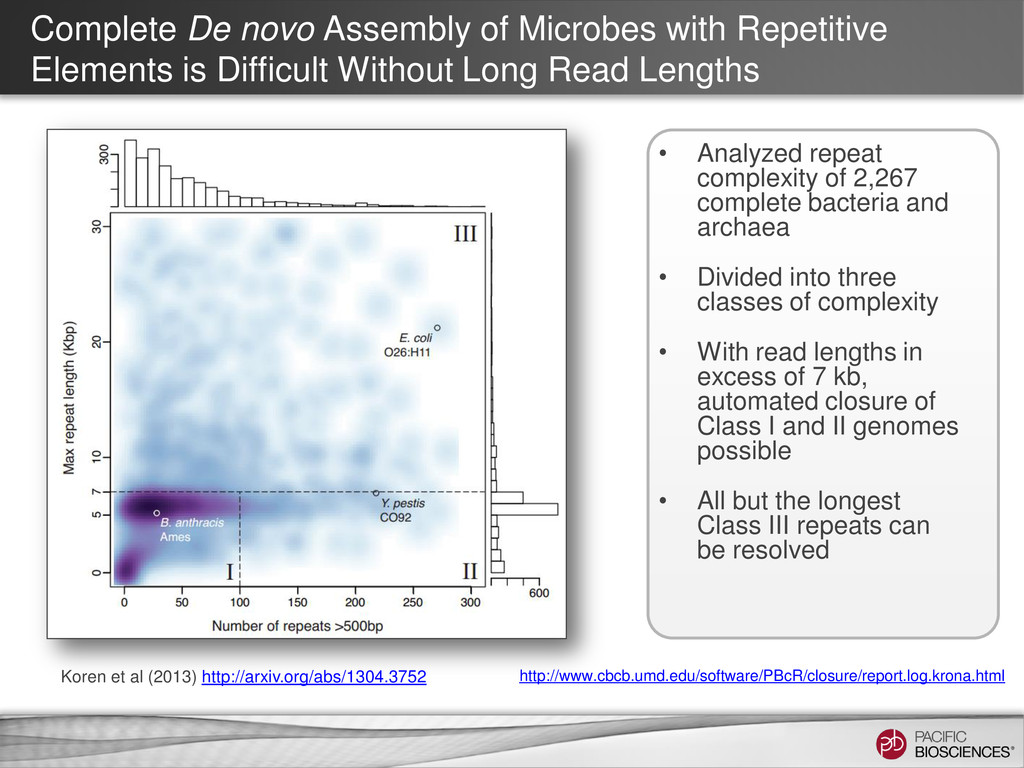
Difficult Without Long Read Lengths • Analyzed repeat complexity of 2,267 complete bacteria and archaea • Divided into three classes of complexity • With read lengths in excess of 7 kb, automated closure of Class I and II genomes possible • All but the longest Class III repeats can be resolved Koren et al (2013) http://arxiv.org/abs/1304.3752 http://www.cbcb.umd.edu/software/PBcR/closure/report.log.krona.html
Sequencing reads with short reads for error correction • Requires multiple types of data, (at least) two library preps, different sequencing technologies…
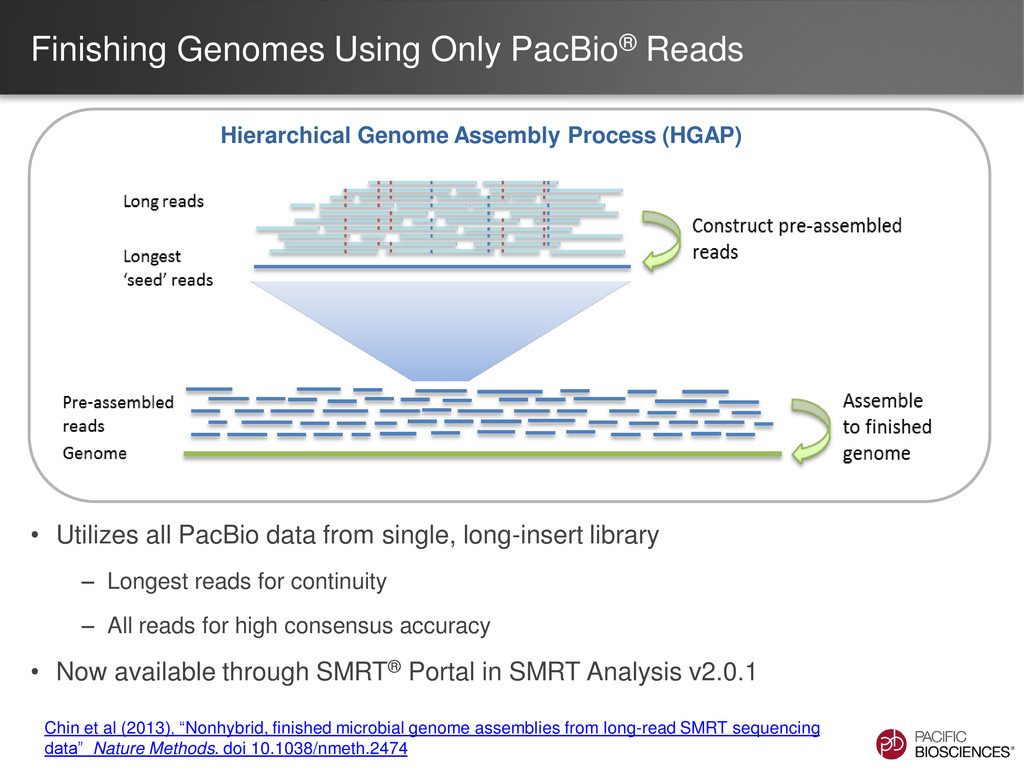
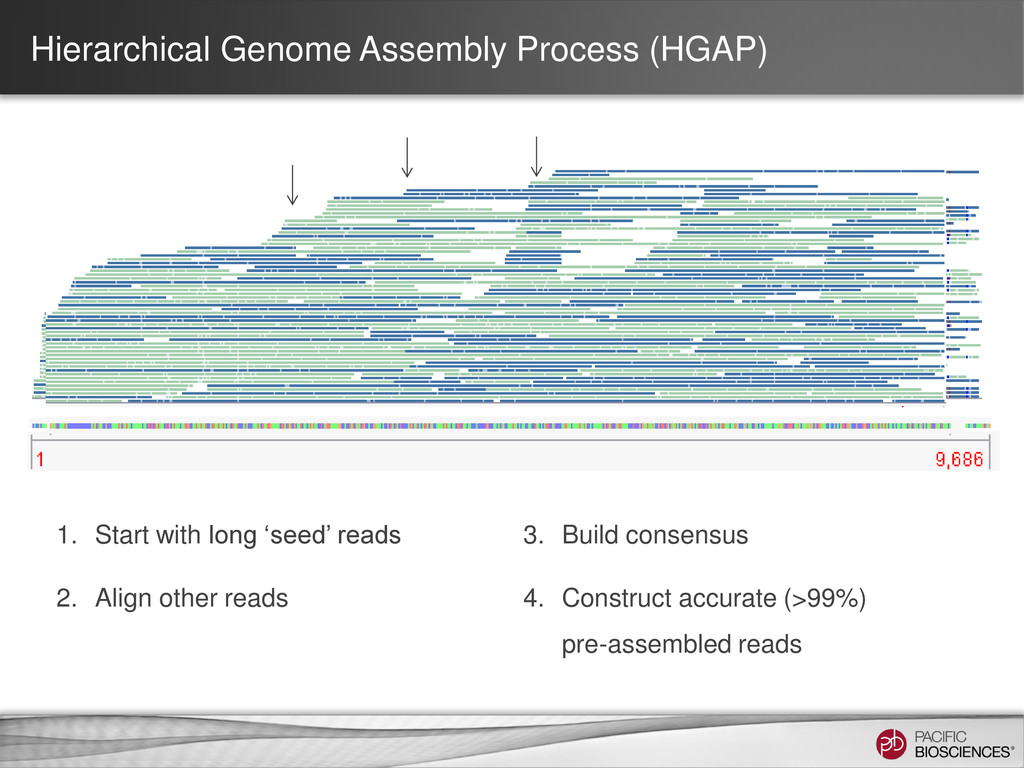
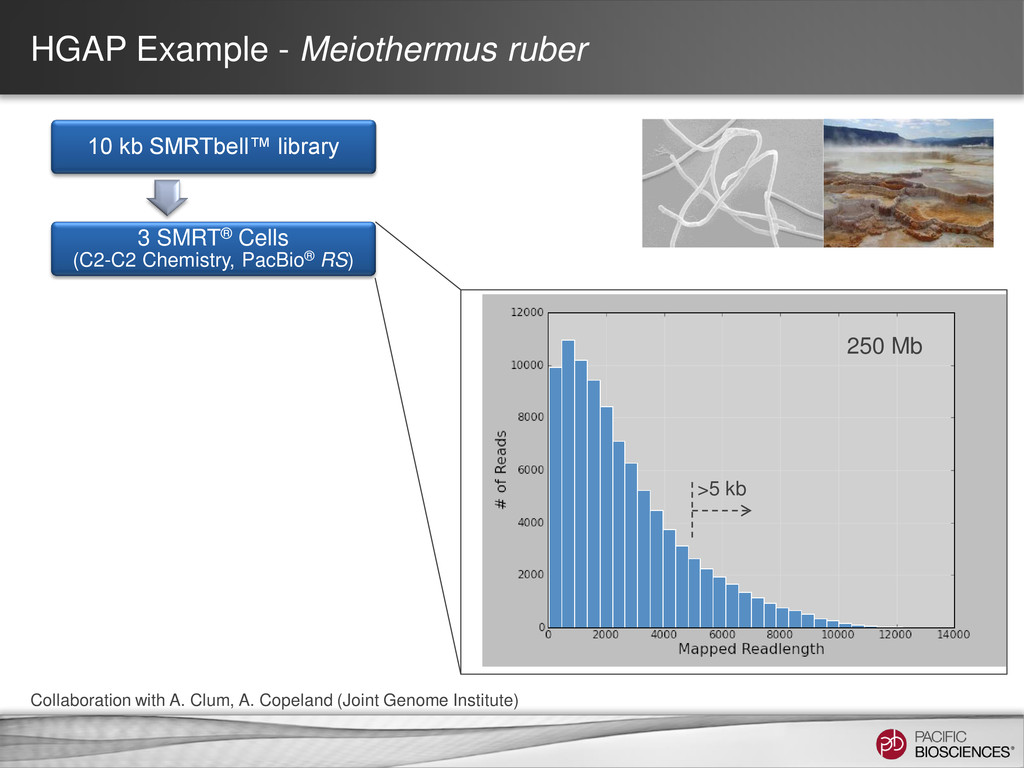
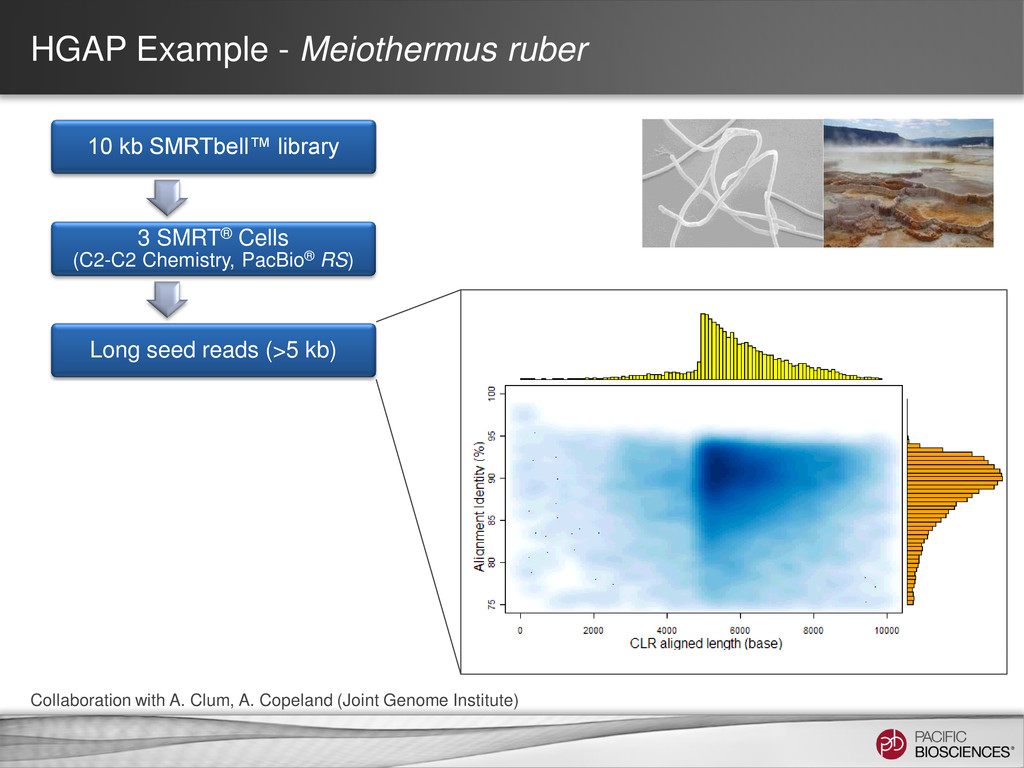
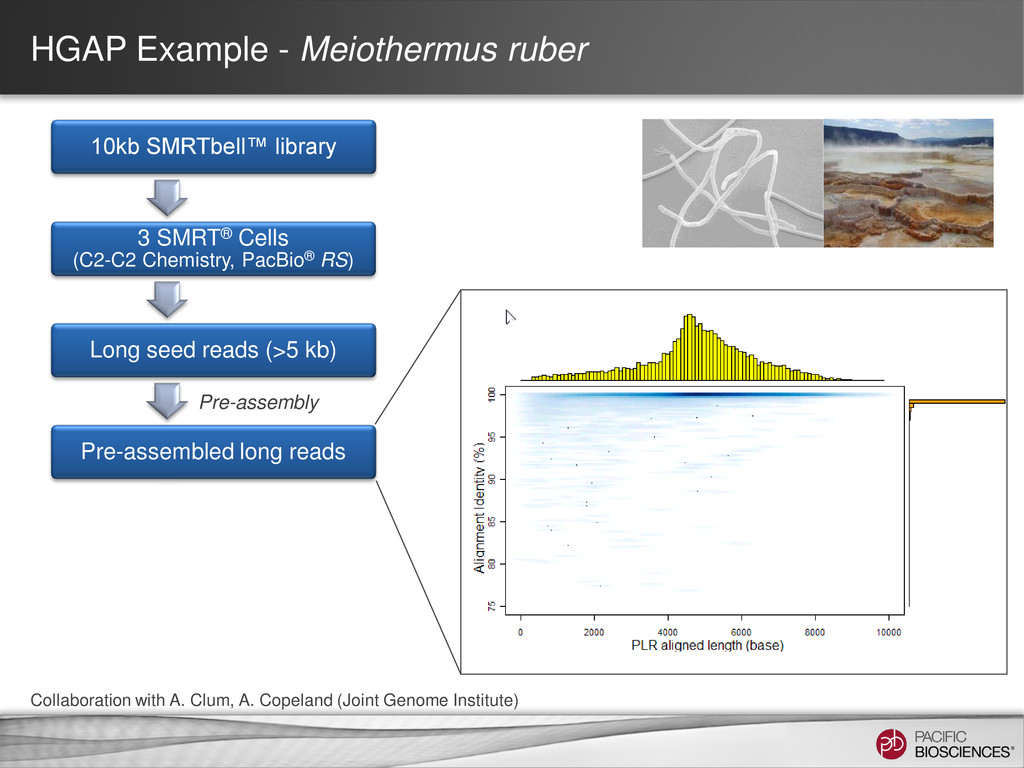
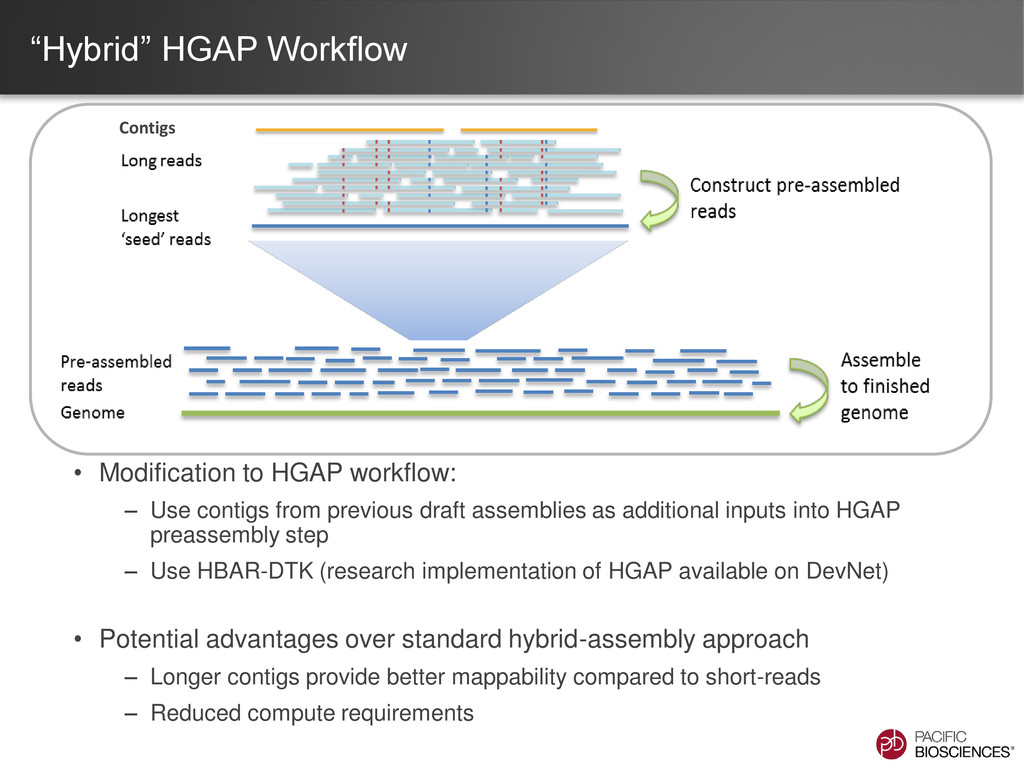
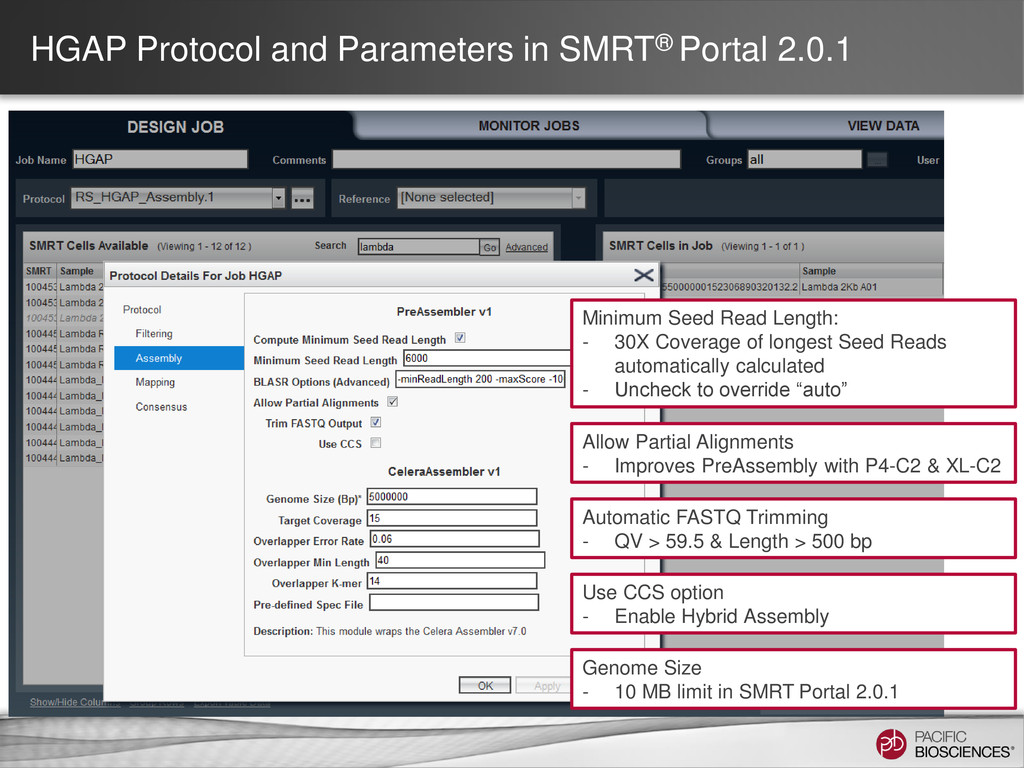
data from single, long-insert library – Longest reads for continuity – All reads for high consensus accuracy • Now available through SMRT® Portal in SMRT Analysis v2.0.1 Hierarchical Genome Assembly Process (HGAP) Chin et al (2013), “Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data” Nature Methods. doi 10.1038/nmeth.2474
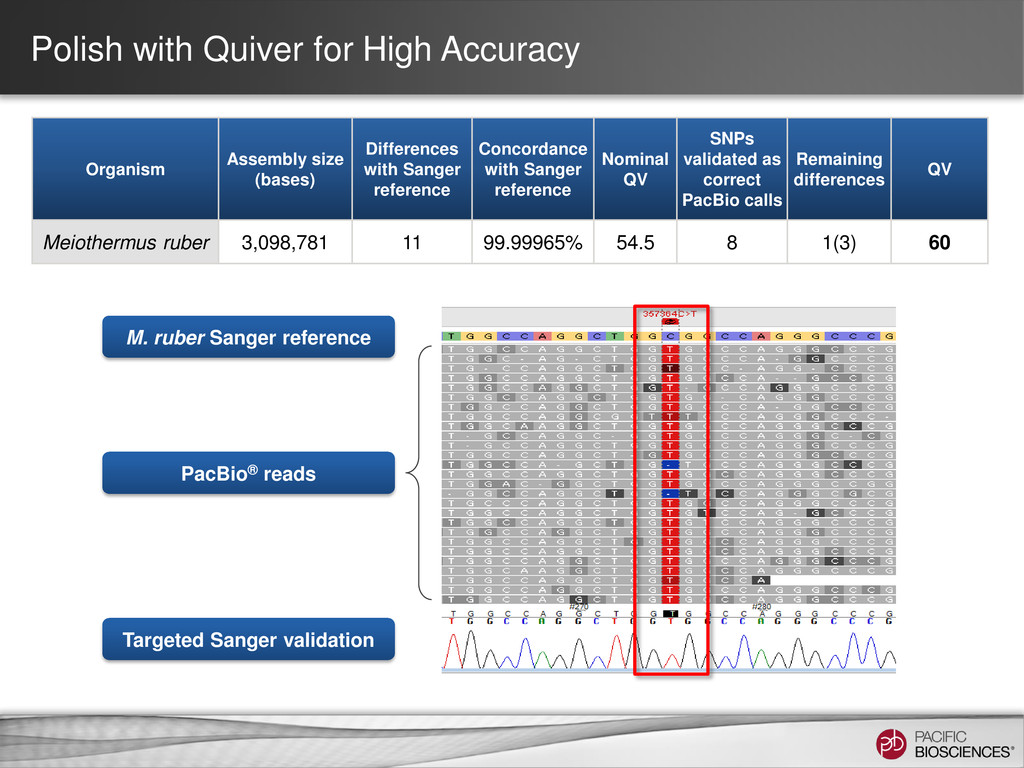
achieve accuracy >Q50 (i.e. > 99.999%) using only PacBio reads • How Quiver works – Takes multiple reads of a given DNA template, outputs best guess of template’s identity – QV-aware hidden Markov model to account for sequencing errors; a greedy algorithm to find the maximum likelihood template – Similar underlying algorithm currently used for CCS generation • Links: – www.pacbiodevnet.com/quiver – https://github.com/PacificBiosciences/GenomicConsensus 15 Quiver Aligned Reads Reference Consensus fasta/fastq Variants.gff
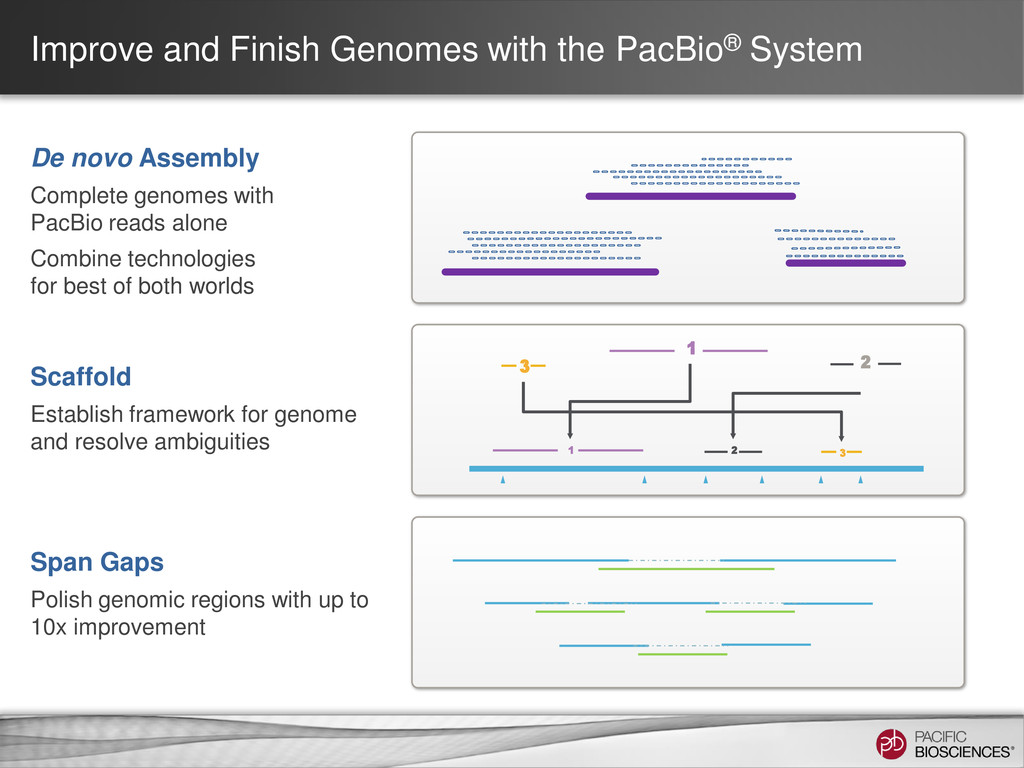
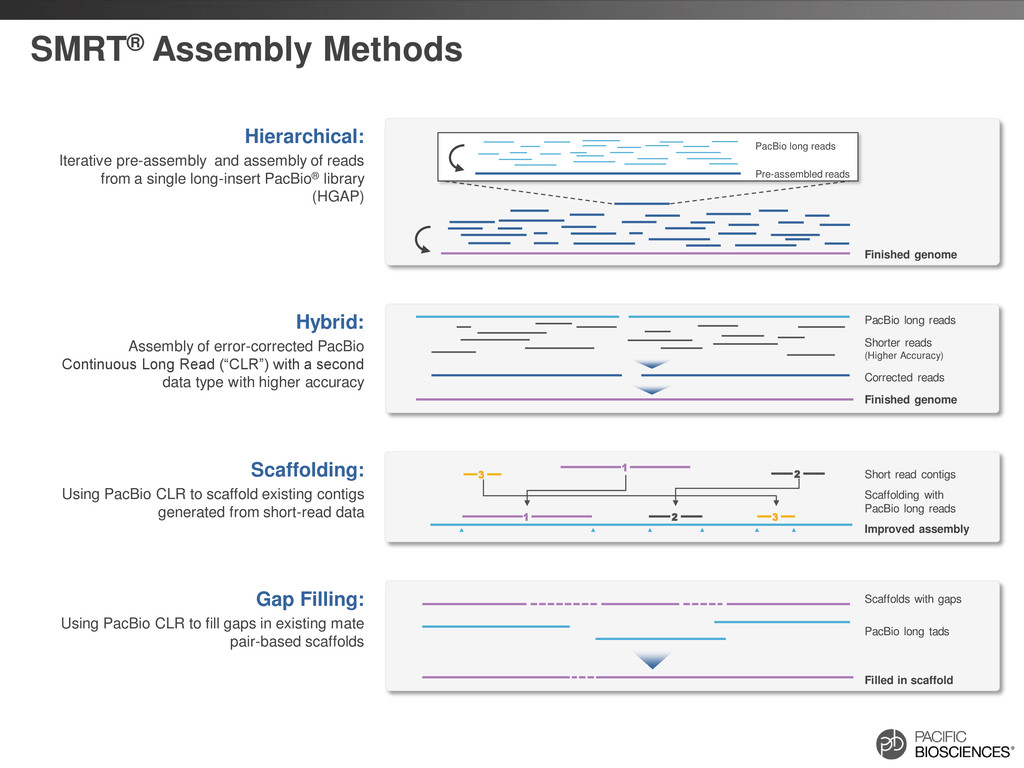
Assembly Complete genomes with PacBio reads alone Combine technologies for best of both worlds 2 3 2 3 1 1 Scaffold Establish framework for genome and resolve ambiguities Span Gaps Polish genomic regions with up to 10x improvement
contigs from previous draft assemblies as additional inputs into HGAP preassembly step – Use HBAR-DTK (research implementation of HGAP available on DevNet) • Potential advantages over standard hybrid-assembly approach – Longer contigs provide better mappability compared to short-reads – Reduced compute requirements Contigs
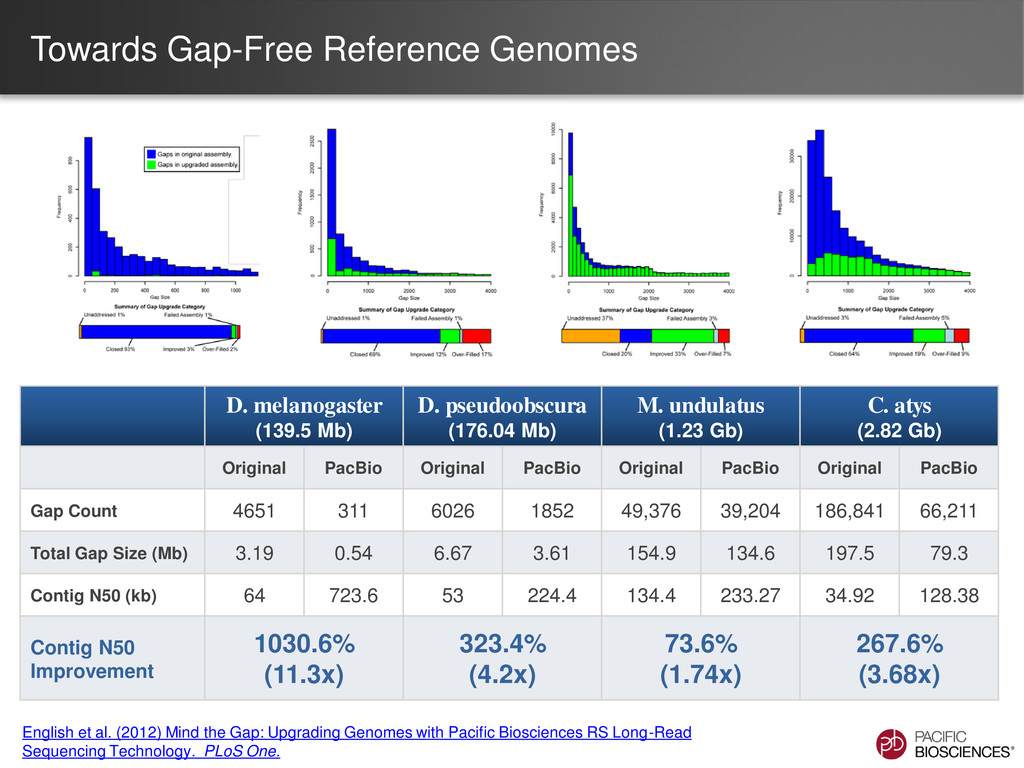
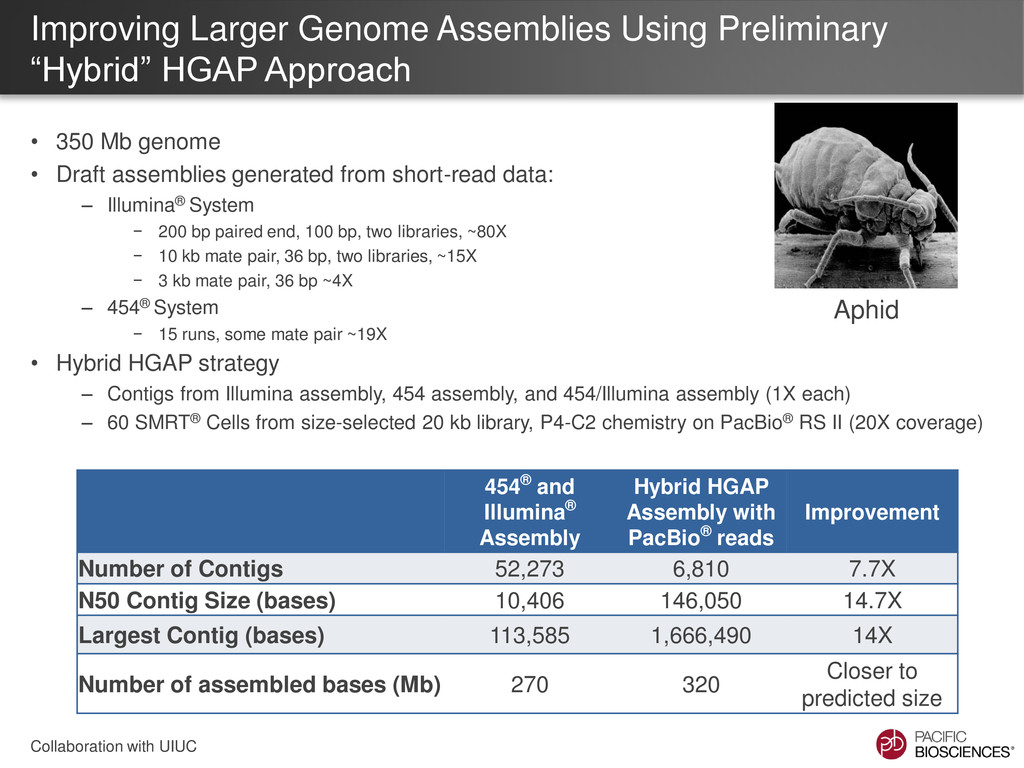
GeneID exons / gene Mean exon size (bp) Mean protein size (aa) Initial Illumina®/454® Assemblies 10.4 2.16 170 123 Hybrid HGAP (PacBio® + Illumina®/454® contigs) 146 3.24 217 284 Pea aphid (Acyr 2.0) 27 2.84 223 212 • GeneID using an aphid model was run on assemblies to predict gene content • Increased continuity of hybrid HGAP assembly yields more exons / gene and higher average protein size • These statistics are more consistent with the published pea aphid assembly, and possibly even better Collaboration with UIUC
RS, the contigs from our de novo assembly of the 400Mbp rice genome are several fold better than the state-of-the- art ALLPATHS-LG assembly using short reads” Michael C. Schatz, Ph.D. Assistant Professor of Quantitative Biology Cold Spring Harbor Laboratory Rice Genome Assembly (Oryza sativa pv Nipponbare: 400 MB) Contig N50 HiSeq® Fragments 50x 2x100bp @ 180 3,925 MiSeq® Fragments 23x 459bp 8x 2x251bp @ 450 6,332 Illumina® Mates 50x 2x100bp @ 180 36x 2x50bp @ 2100 51x 2x50bp @ 4800 18,248 PBeCR + Illumina® Mates 7x 3500bp ** MiSeq for correction 50,995 PBeCR + Illumina® Mates 19x ** MiSeq for correction 155 kb Case Study: The Next Frontier in Assembly – Long Reads Offer Finished Genomes
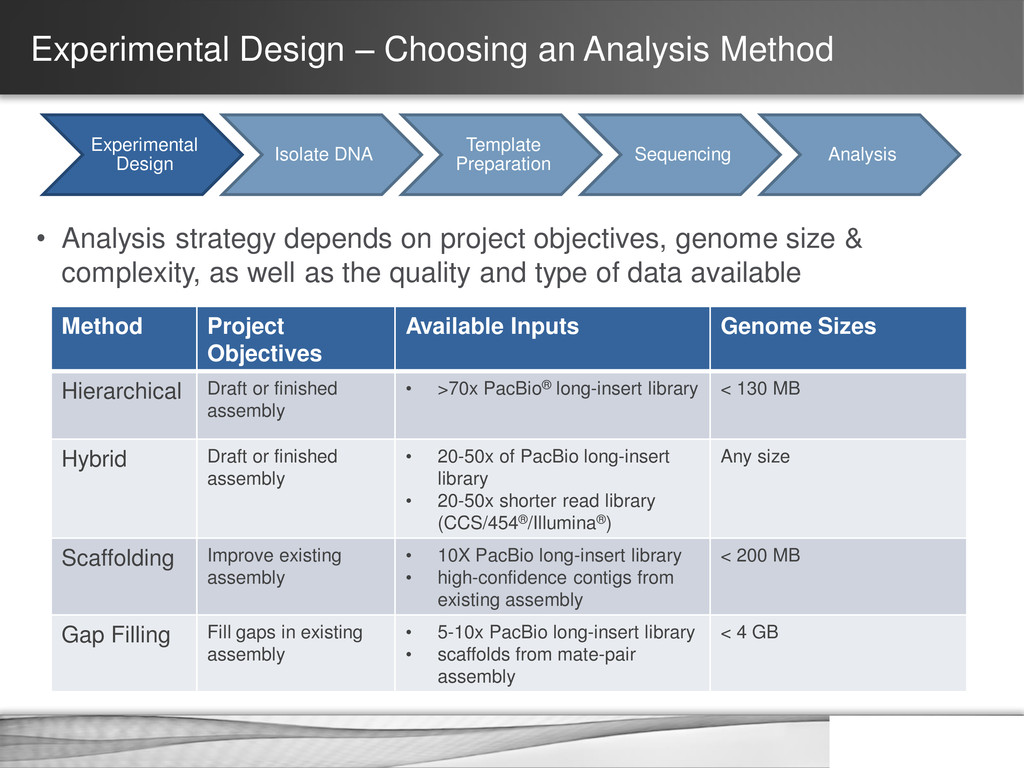
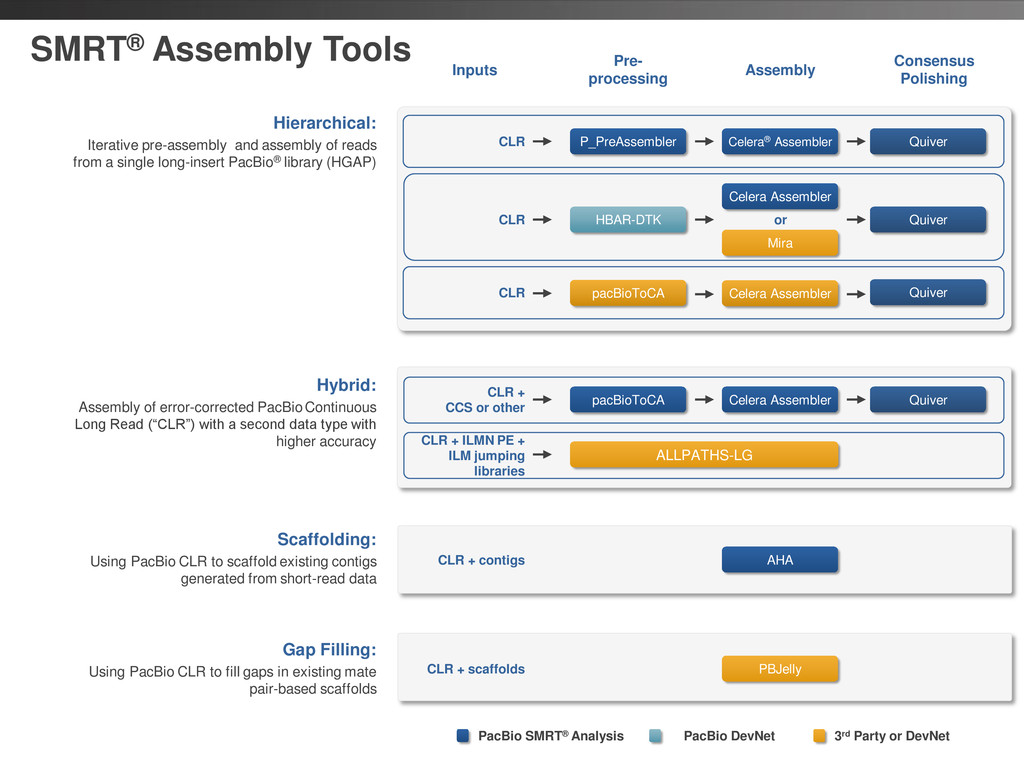
DNA Template Preparation Sequencing Analysis • Analysis strategy depends on project objectives, genome size & complexity, as well as the quality and type of data available Method Project Objectives Available Inputs Genome Sizes Hierarchical Draft or finished assembly • >70x PacBio® long-insert library < 130 MB Hybrid Draft or finished assembly • 20-50x of PacBio long-insert library • 20-50x shorter read library (CCS/454®/Illumina®) Any size Scaffolding Improve existing assembly • 10X PacBio long-insert library • high-confidence contigs from existing assembly < 200 MB Gap Filling Fill gaps in existing assembly • 5-10x PacBio long-insert library • scaffolds from mate-pair assembly < 4 GB
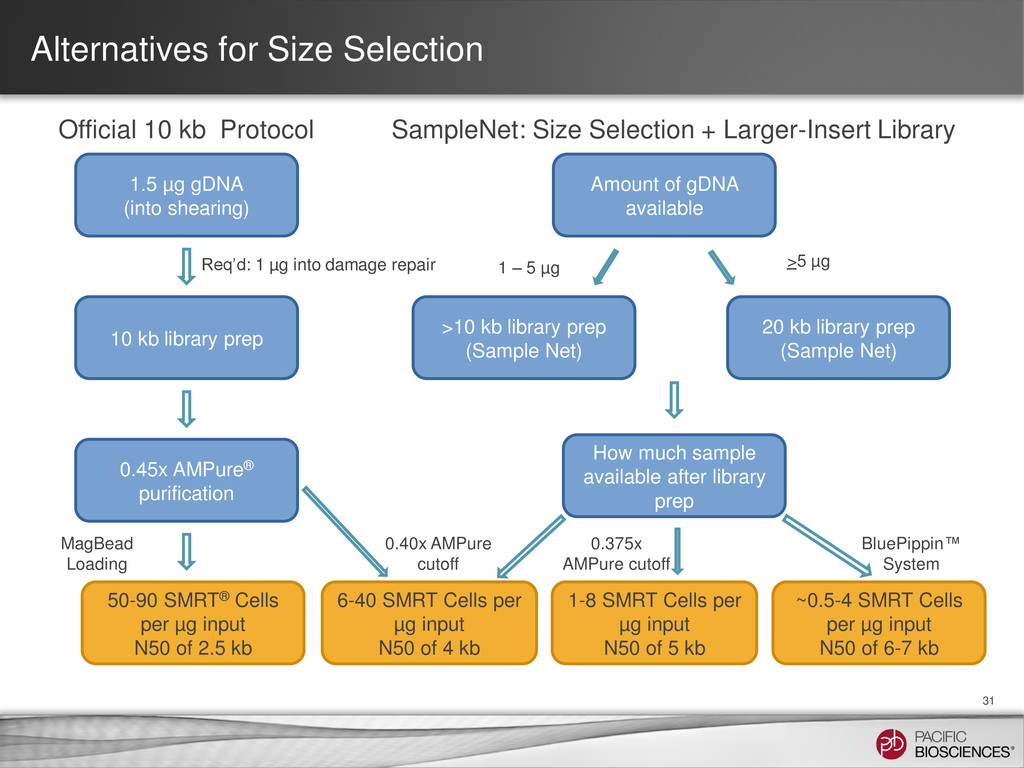
to maximize potential performance • No amplification step during library preparation • Recommendations: – Take care during extraction to avoid gDNA damage & avoid contaminants – Use extraction methods or kits that produce very high molecular weight gDNA – If contaminants are present, purify starting DNA material prior to library prep – Accurately quantify and qualitatively evaluate gDNA – Include DNA damage repair step in library prep 28 Experimental Design Isolate DNA Template Preparation Sequencing Analysis
• Double-stranded DNA Sample (dsDNA) • Minimized freeze-thaw cycles • No exposure to high temp (>65° C) • No exposure to pH extremes (<6 or >9) • Minimize gDNA vortexing and pipetting; pipette gently • OD260/280 between 1.8 and 2.0 • OD260/230 between 2.0 and 2.2 • No insoluble material • No RNA contamination • No exposure to UV or intercalating fluorescent dyes • No chelating agents, divalent metal cations, denaturants, or detergents • No carryover contamination (e.g. polysaccharides) from starting organism 29
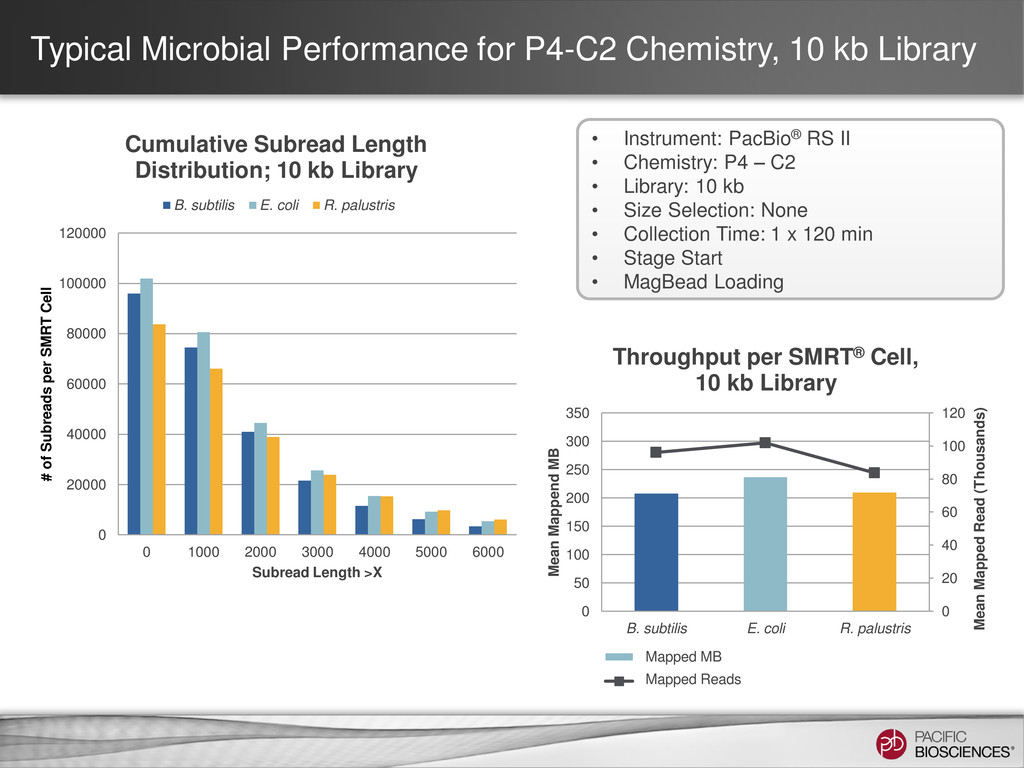
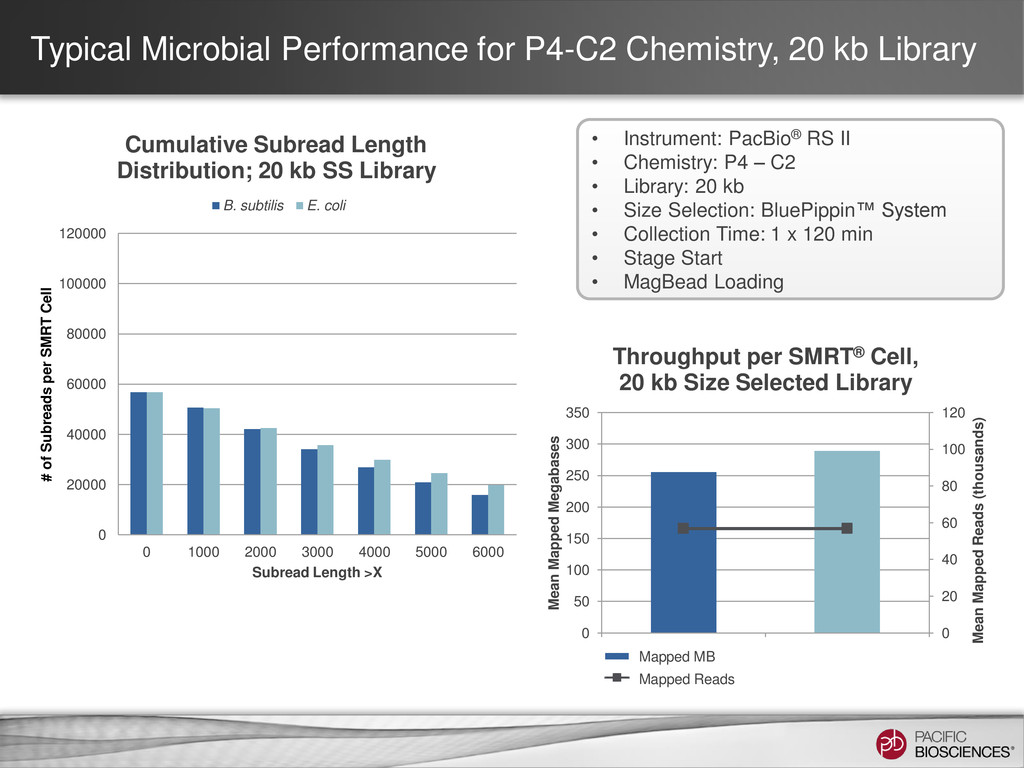
is highly dependent on sample quality & library insert size • Potential sources of variability – Sample damage – Sample degradation – Contaminants – Shearing size & distribution • Important to QC sample if performing size section • Use of XL DNA Sequencing Kit 1.0 (P4 pol + XL Seq Kit) with known low- quality or short-insert libraries is Not Recommended – Unlikely to see subread length gain compared to P4 Binding Kit + C2 Sequencing Kit condition (P4-C2), but will see a drop in consensus accuracy & throughput 33
P4 Enzyme for High Accuracy & Long Read Lengths • For most applications where consensus accuracy matters, would recommend combining P4 Binding kit with DNA Sequencing Kit (P4 – C2) • Optional XL DNA Sequencing Kit – Slightly longer read lengths, but at a cost of consensus accuracy – Better suited for scaffolding, spanning structural rearrangements, spanning long repeats, etc. 36 DNA Template Prep Kits • DNA Template Prep Kit 2.0 (250 bp ‒ <3 kb) • DNA Template Prep Kit 2.0 (3 kb ‒ <10 kb) DNA/Polymerase Binding Kit • New: DNA/Polymerase Binding Kit P4 DNA Sequencing Kit • DNA Sequencing Kit 2.0 • XL DNA Sequencing Kit 1.0 (Optional)
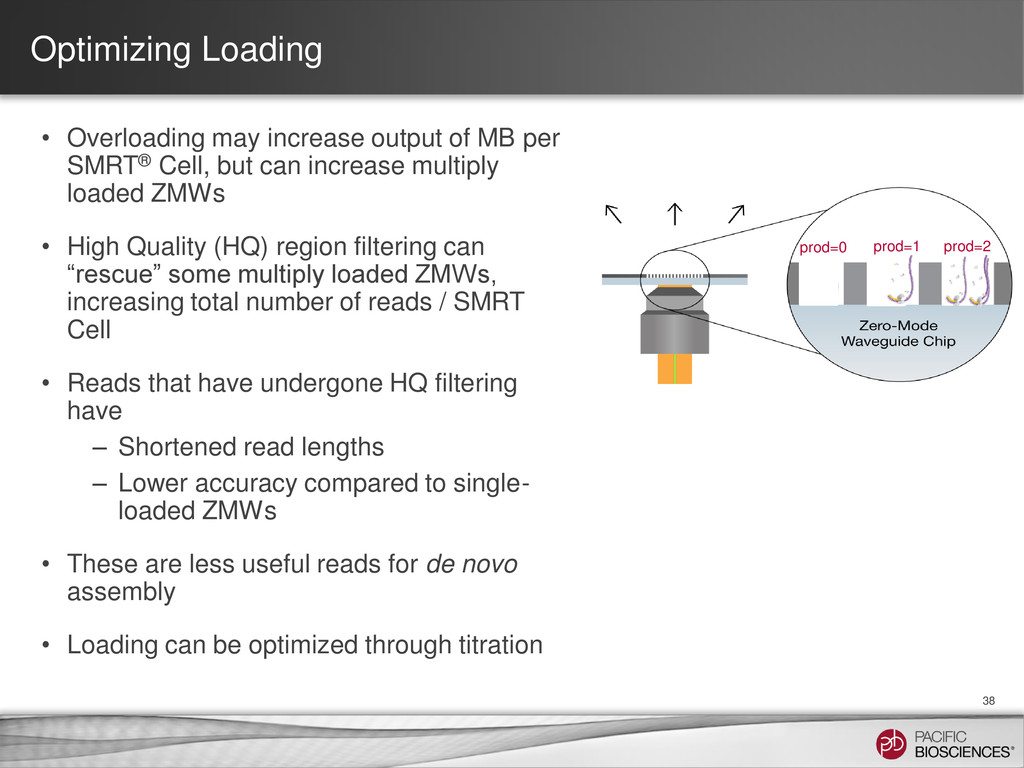
SMRT® Cell, but can increase multiply loaded ZMWs • High Quality (HQ) region filtering can “rescue” some multiply loaded ZMWs, increasing total number of reads / SMRT Cell • Reads that have undergone HQ filtering have – Shortened read lengths – Lower accuracy compared to single- loaded ZMWs • These are less useful reads for de novo assembly • Loading can be optimized through titration 38 prod=0 prod=1 prod=2
Hierarchical: Iterative pre-assembly and assembly of reads from a single long-insert PacBio® library (HGAP) Scaffolds with gaps PacBio long tads Filled in scaffold Gap Filling: Using PacBio CLR to fill gaps in existing mate pair-based scaffolds Scaffolding with PacBio long reads Improved assembly Short read contigs Scaffolding: Using PacBio CLR to scaffold existing contigs generated from short-read data 2 3 2 3 1 1 PacBio long reads Shorter reads (Higher Accuracy) Finished genome Hybrid: Assembly of error-corrected PacBio Continuous Long Read (“CLR”) with a second data type with higher accuracy Corrected reads
CLR + contigs PBJelly CLR + scaffolds PacBio SMRT® Analysis PacBio DevNet 3rd Party or DevNet HBAR-DTK P_PreAssembler Celera Assembler Mira Quiver CLR or Quiver pacBioToCA ALLPATHS-LG Celera Assembler CLR + CCS or other CLR + ILMN PE + ILM jumping libraries Hybrid: Assembly of error-corrected PacBio Continuous Long Read (“CLR”) with a second data type with higher accuracy Hierarchical: Iterative pre-assembly and assembly of reads from a single long-insert PacBio® library (HGAP) Scaffolding: Using PacBio CLR to scaffold existing contigs generated from short-read data Gap Filling: Using PacBio CLR to fill gaps in existing mate pair-based scaffolds pacBioToCA Celera Assembler Quiver CLR CLR Quiver Celera® Assembler
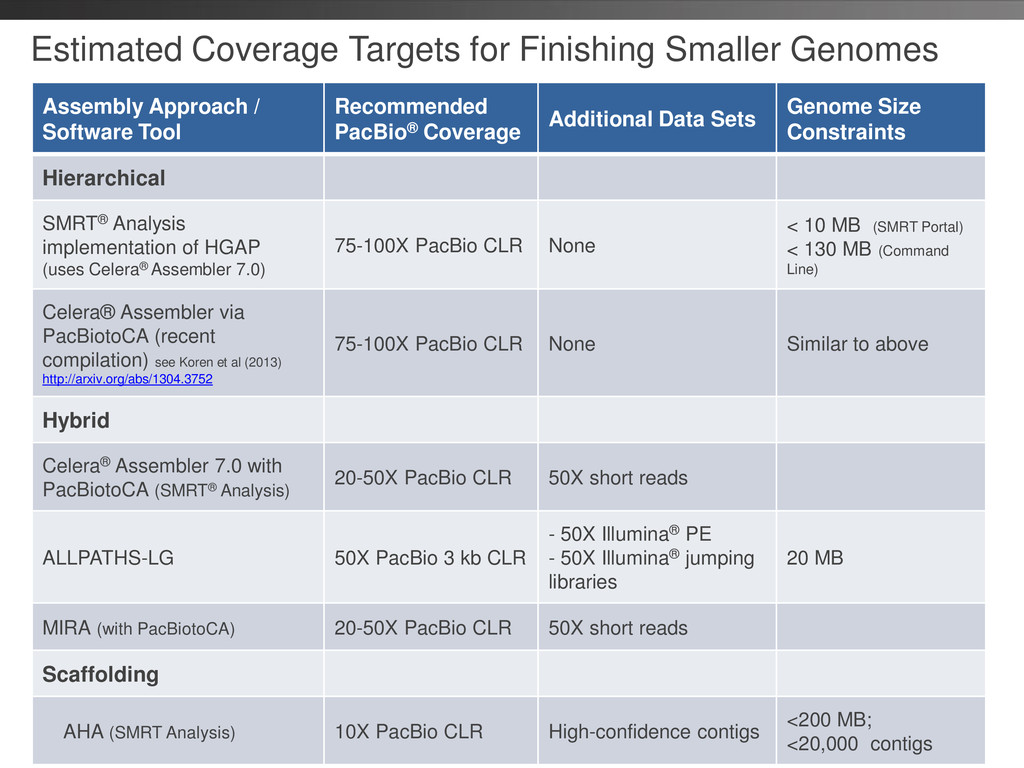
critical to lower read coverage requirements • Total single-pass coverage for best results: > 70X • SMRT® Portal imposed 10 MB genome size limit (safely within our min compute requirements) • SMRT Pipe HGAP tested to 100 MB genome size (4 GB seed read limit) – Larger genomes can be processed with HGAP Developer Kit (DevNet) BUT this has not been extensively validated • Additional/latest information can be found in HGAP wiki or by contacting Field BFX Group https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/HGAP 44 Watch Training Video: Bacterial Assembly and Epigenetic Analysis
on BFX wiki site • Modularized for even more control: – Run P_PreAssembler with SMRT® Pipe on the command-line – Run Celera® Assembler on the command-line – Use Quiver option of P_GenomicConsensus to polish the assembly – Additional tweaks to filtering and trimming may improve assembly More details here: https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/HGAP https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/HGAP-2.0 47
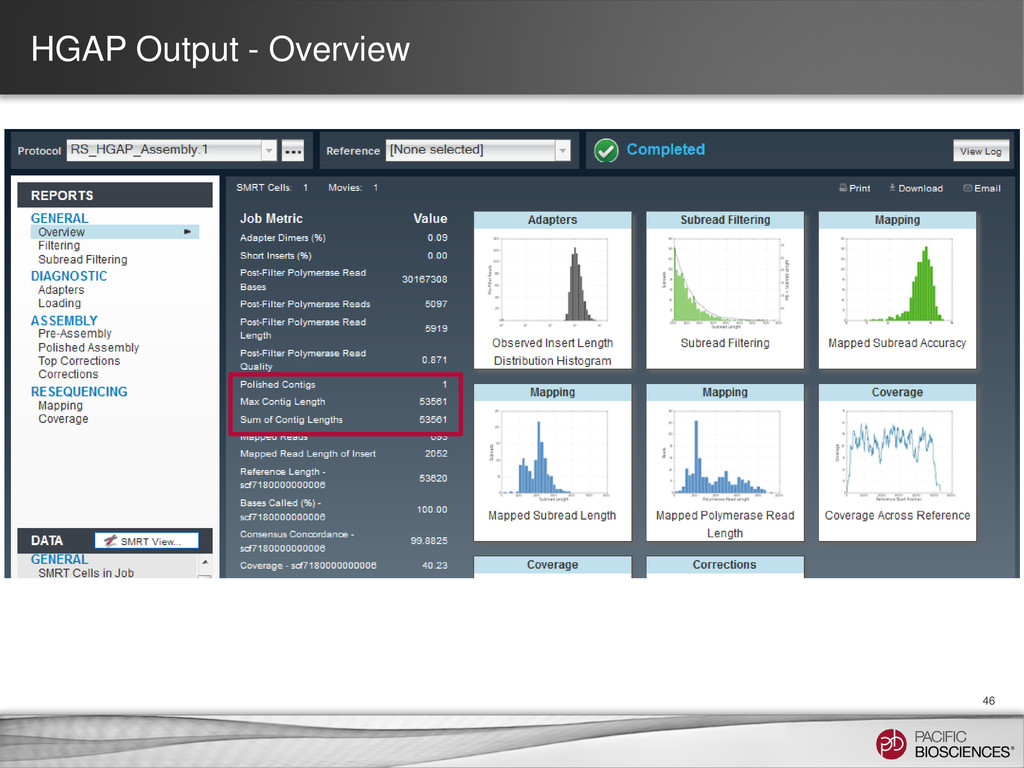
of contigs – N50: Equal to the size of the contig found if you sort contigs by size and walk to the contig that represents 50% of the total sequence − N50 = 10 bp − Mean contig length = 3 bp – Max contig size • Limitation of these metrics: – They do not capture information about assembly accuracy! − Large scale mis-assemblies − Base level errors – There might be more than one chromosome (plasmid, phage, etc.) – Contaminants may contribute to a contig (such as a cloning vector) 48 10 4 1 1 1 1
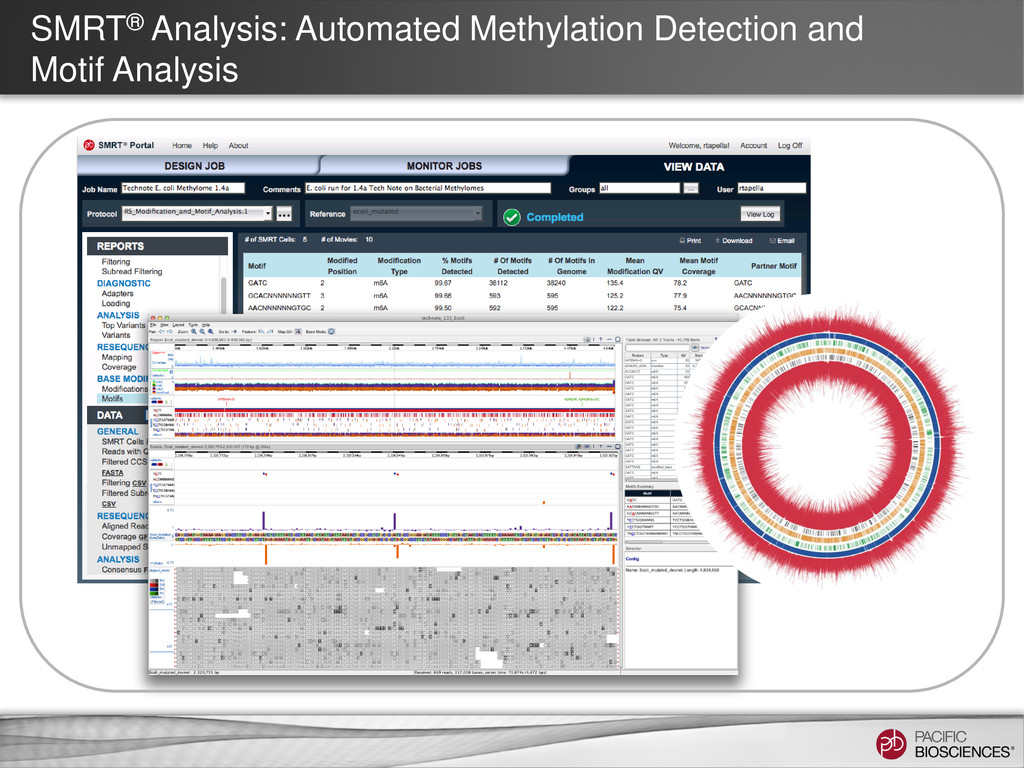
Assembly Results – Ensure minimum coverage and subread length thresholds met – Check for coverage uniformity − Spikes/valleys evidence of mis-assemblies − Low coverage, short contigs may be discarded – Look for evidence of plasmids in degenerates file – Ensure at least 90% of reads are mapping to assembly – Evaluate circularity of chromosomes and plasmids (Gepard) • Additional Ways to Improve Final Assembly – Parameter optimization of HGAP – Manual trimming of ends may be needed for circular genomes – Minimus2 and AHA to join contigs • Post Assembly Analysis – Methylation Detection and Motif Analysis) – Phage insertions (PHAST (http://phast.wishartlab.com) 49 https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/Finishing-Bacterial-Genomes
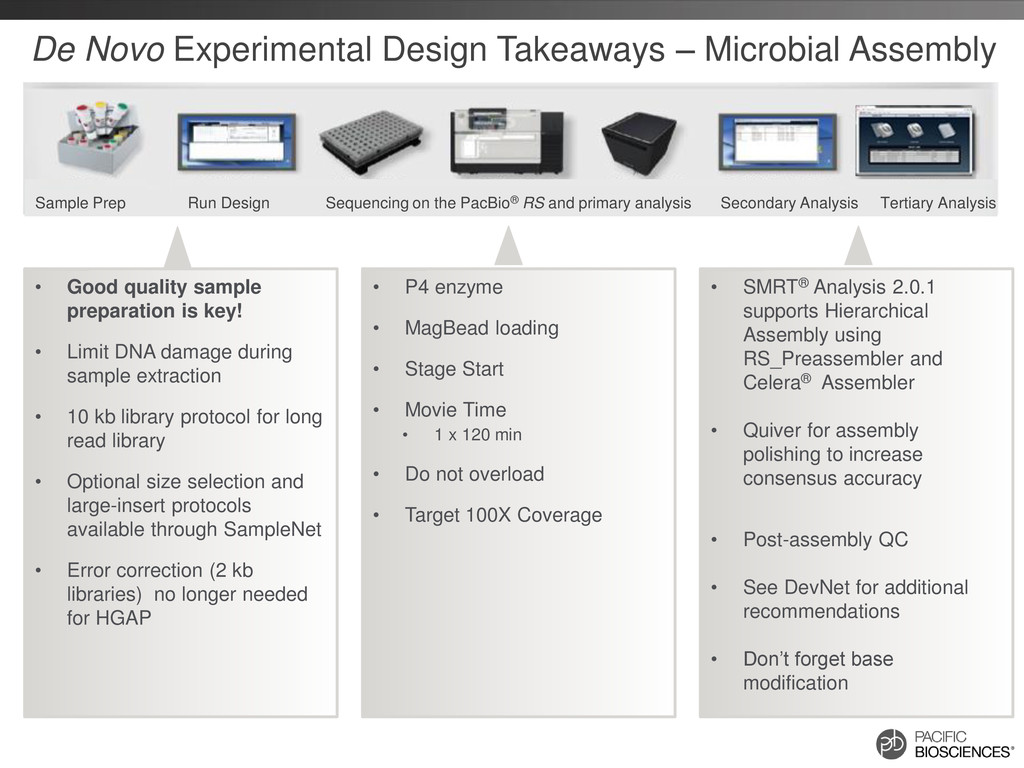
enzyme • MagBead loading • Stage Start • Movie Time • 1 x 120 min • Do not overload • Target 100X Coverage • SMRT® Analysis 2.0.1 supports Hierarchical Assembly using RS_Preassembler and Celera® Assembler • Quiver for assembly polishing to increase consensus accuracy • Post-assembly QC • See DevNet for additional recommendations • Don’t forget base modification • Good quality sample preparation is key! • Limit DNA damage during sample extraction • 10 kb library protocol for long read library • Optional size selection and large-insert protocols available through SampleNet • Error correction (2 kb libraries) no longer needed for HGAP Sample Prep Run Design Sequencing on the PacBio® RS and primary analysis Secondary Analysis Tertiary Analysis
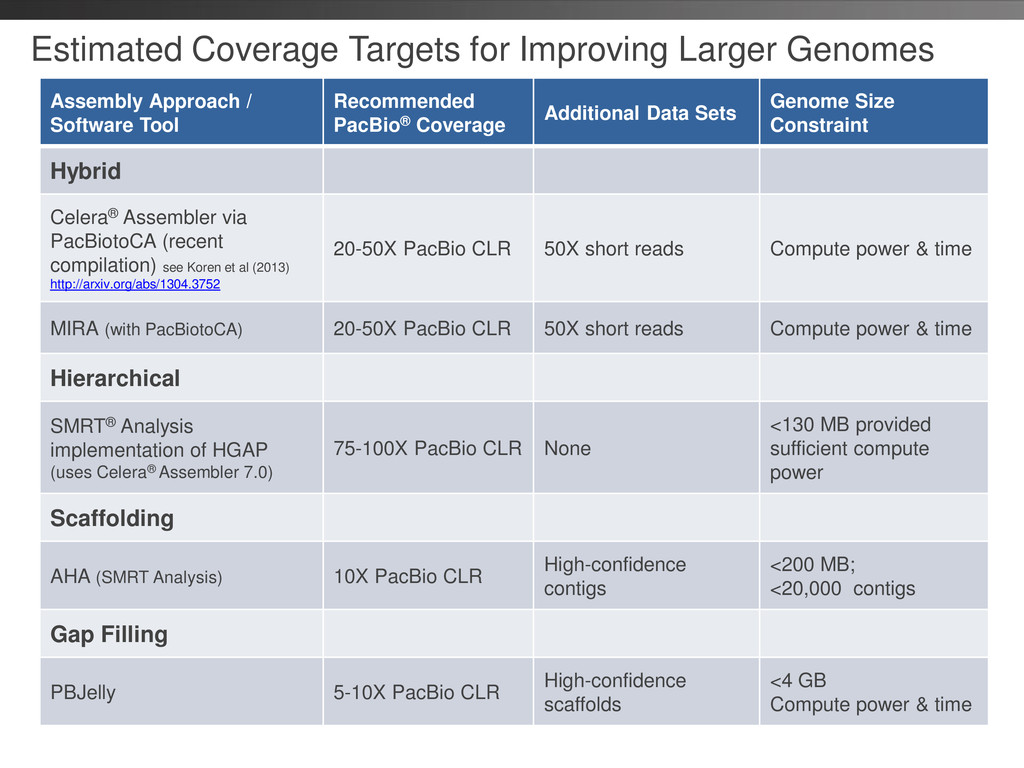
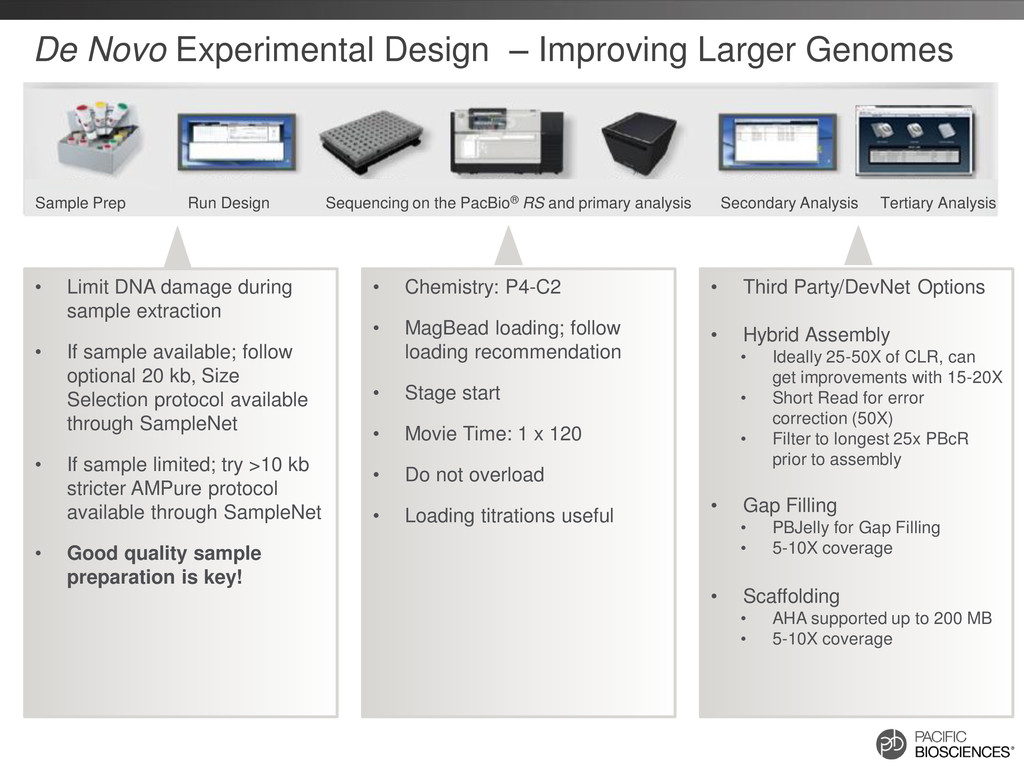
P4-C2 • MagBead loading; follow loading recommendation • Stage start • Movie Time: 1 x 120 • Do not overload • Loading titrations useful • Third Party/DevNet Options • Hybrid Assembly • Ideally 25-50X of CLR, can get improvements with 15-20X • Short Read for error correction (50X) • Filter to longest 25x PBcR prior to assembly • Gap Filling • PBJelly for Gap Filling • 5-10X coverage • Scaffolding • AHA supported up to 200 MB • 5-10X coverage • Limit DNA damage during sample extraction • If sample available; follow optional 20 kb, Size Selection protocol available through SampleNet • If sample limited; try >10 kb stricter AMPure protocol available through SampleNet • Good quality sample preparation is key! Sample Prep Run Design Sequencing on the PacBio® RS and primary analysis Secondary Analysis Tertiary Analysis
of presentations, posters and other de novo assembly resources available through PacBio’s website (www.pacb.com/denovo) • Protocols, Technical & Application Notes available through Customer Portal • DevNet – HGAP Reference Implementation: http://www.smrtcommunity.com/Share/Code?id=a1q70000000H2qRAAS – Quiver: www.pacbiodevnet.com/quiver – Bacterial Assembly and Epigenetic Analysis Training Web Video http://www.pacificbiosciences.com/Tutorials/Bacterial_Assembly_Epigenetic_Analysis_HGA P/story_html5.html • Additional information on Assembly Tools – Celera® Assembler: http://sourceforge.net/apps/mediawiki/wgs- assembler/index.php?title=PacBioToCA – Allpaths-LG: http://www.broadinstitute.org/software/allpaths-lg/blog/ – PBJelly: http://sourceforge.net/p/pb-jelly/wiki/Home/ 56
are trademarks of Pacific Biosciences in the United States and/or other countries. All other trademarks are the sole property of their respective owners.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}