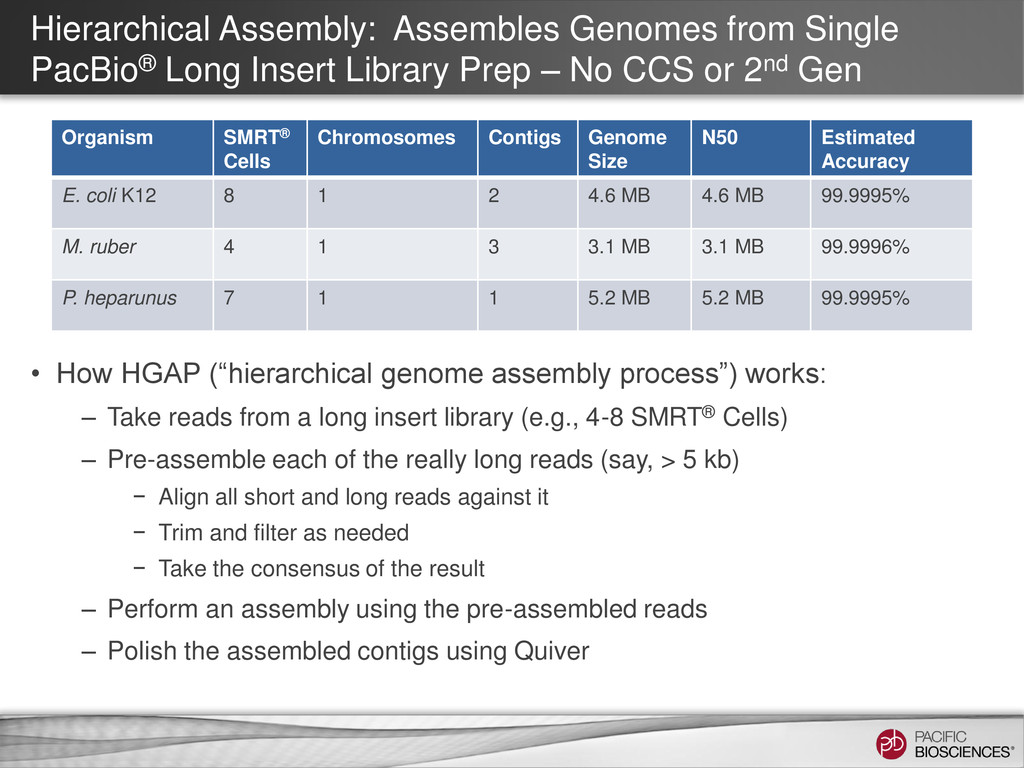
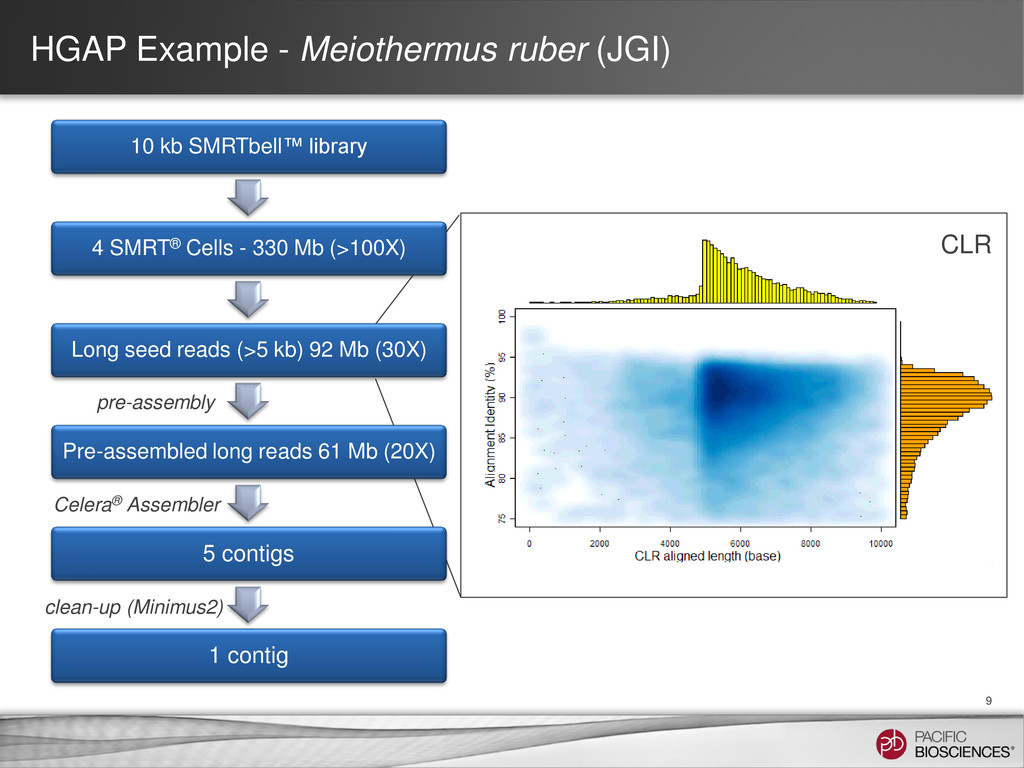
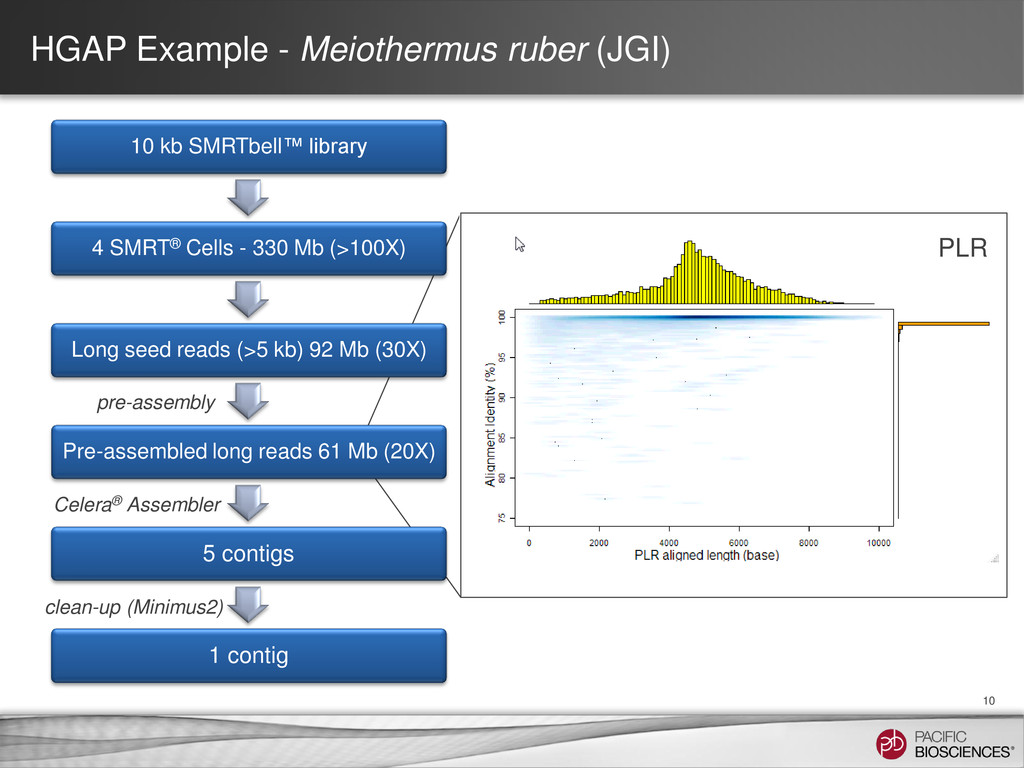
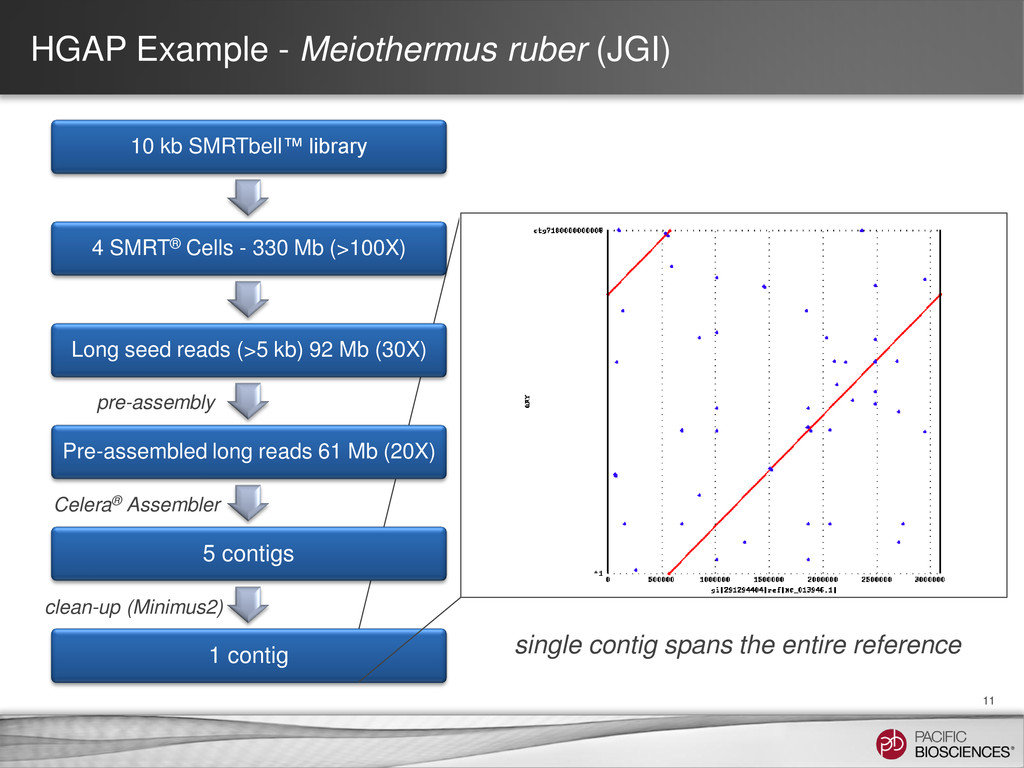
Prep – No CCS or 2nd Gen • How HGAP (“hierarchical genome assembly process”) works: – Take reads from a long insert library (e.g., 4-8 SMRT® Cells) – Pre-assemble each of the really long reads (say, > 5 kb) − Align all short and long reads against it − Trim and filter as needed − Take the consensus of the result – Perform an assembly using the pre-assembled reads – Polish the assembled contigs using Quiver Organism SMRT® Cells Chromosomes Contigs Genome Size N50 Estimated Accuracy E. coli K12 8 1 2 4.6 MB 4.6 MB 99.9995% M. ruber 4 1 3 3.1 MB 3.1 MB 99.9996% P. heparunus 7 1 1 5.2 MB 5.2 MB 99.9995%
• Used reference (Sanger) from the Joint Genome Institute to evaluate assembly concordance • Assessment of initial differences: – QV ~43.4 (99.9954%) – 141 differences between the assembly and the reference 12 actual: variant caller:
evaluate assembly concordance • Assessment of initial differences: – QV ~43.4 (99.9954%) – 141 differences between the assembly and the reference • Final accuracy post-Quiver: – QV ~54.5 (99.99964%) – 11 differences between the assembly and the reference 13 Initial Assessment of M. ruber Assembly from 4 SMRT® Cells
® Analysis 1.4 (via SMRT Portal) P_PreAssembler protocol in SMRT ® Analysis 1.4 (via Command Line) Reference Implementation of HGAP available on DevNet Skill set required General user Command line skill Savvy bioinformatician Genome size BACs or Viral assembly Microbial size Up to 100 MB tested Assembly Performance Fine for small genomes, not recommended and slow for larger genomes Good results, but may require parameter tweaking Good results Installation difficulty Part of SMRT Analysis 1.4 Part of SMRT Analysis 1.4 High (requires compiling code, cluster configuration, etc.) Target user General users who want to try the HGAP workflow and test on a small genome Bioinformatics users new to HGAP Customers already introduced to DevNet HGAP Our recommendation
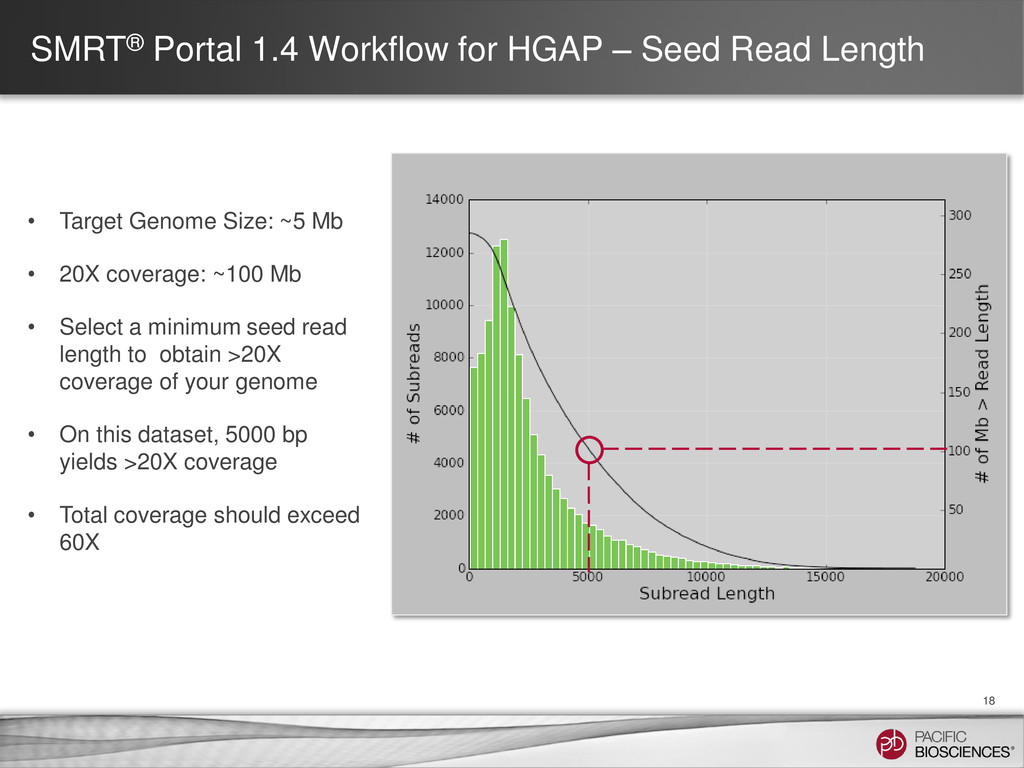
18 • Target Genome Size: ~5 Mb • 20X coverage: ~100 Mb • Select a minimum seed read length to obtain >20X coverage of your genome • On this dataset, 5000 bp yields >20X coverage • Total coverage should exceed 60X
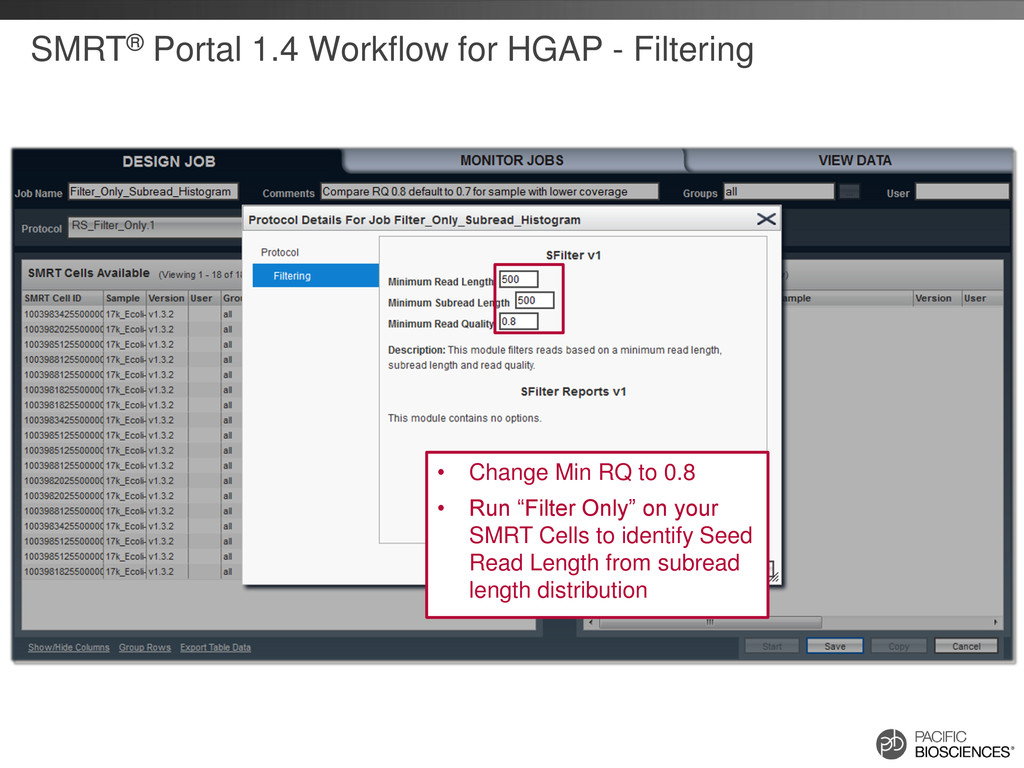
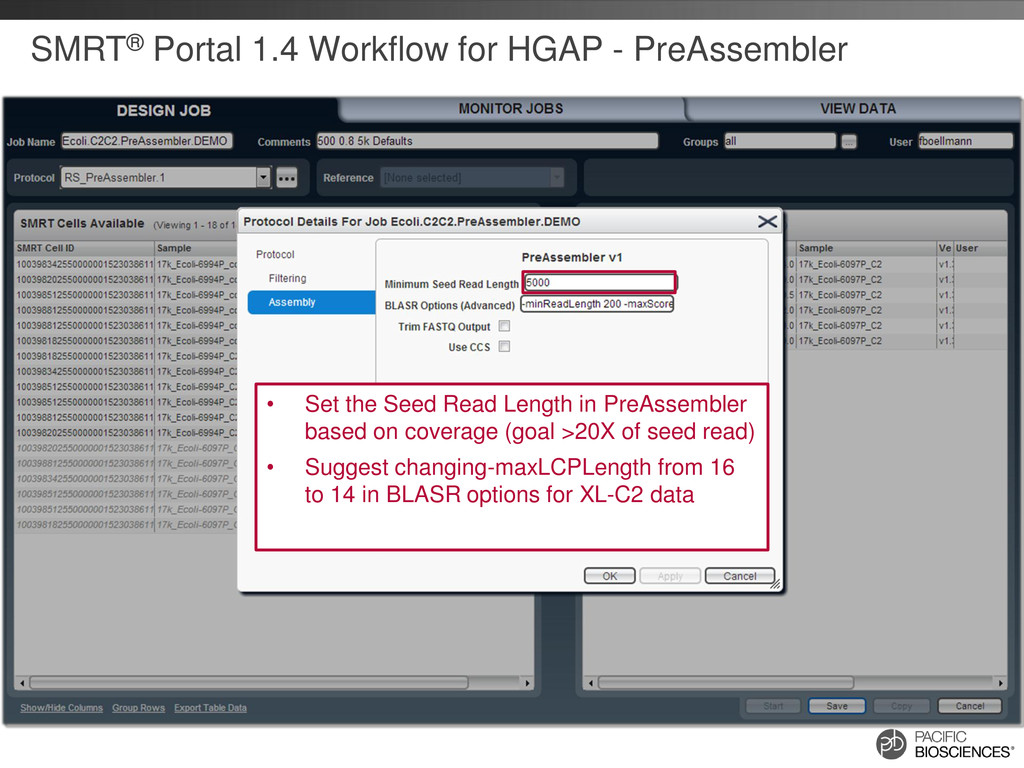
the Seed Read Length in PreAssembler based on coverage (goal >20X of seed read) • Suggest changing-maxLCPLength from 16 to 14 in BLASR options for XL-C2 data
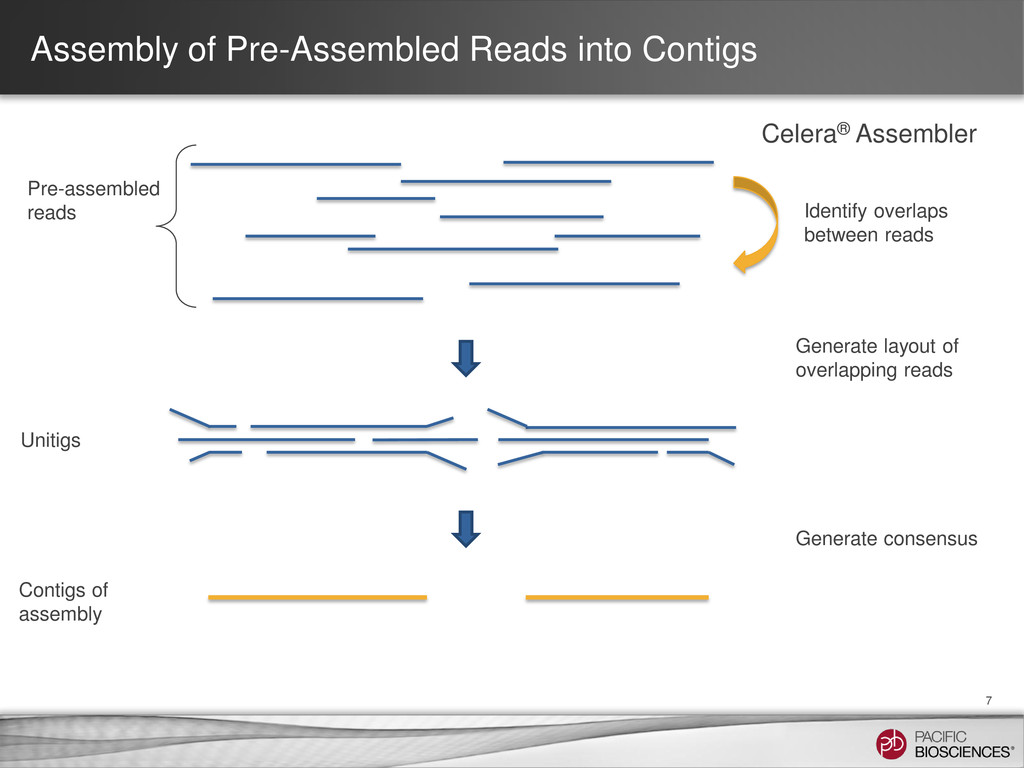
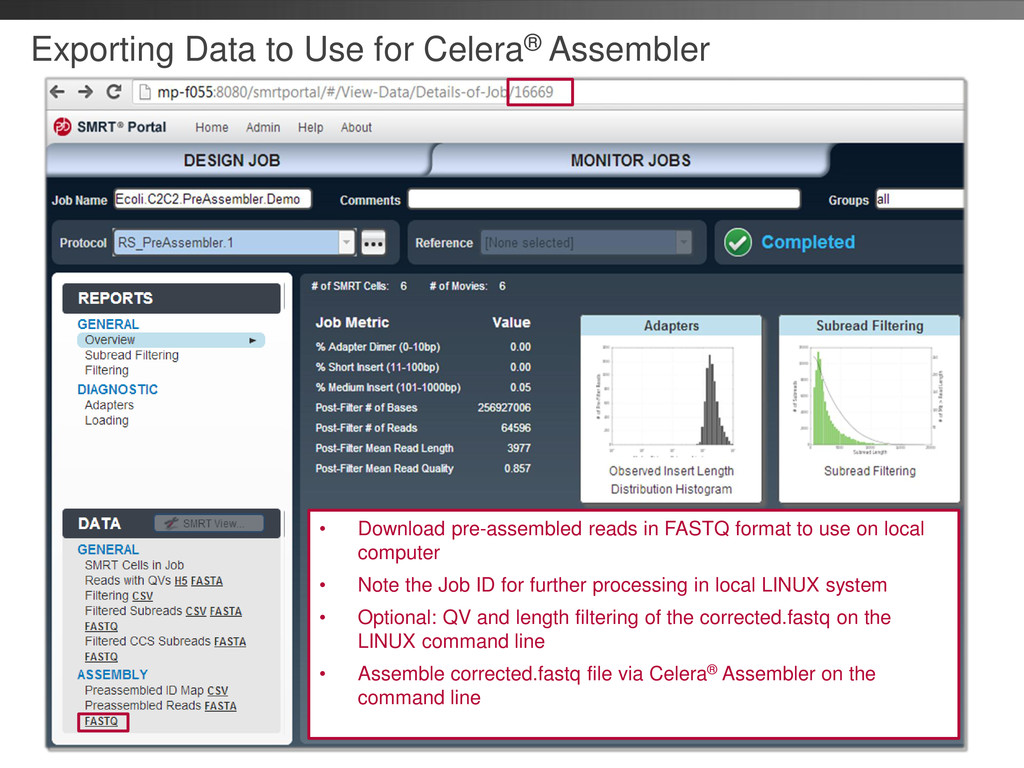
local computer • Note the Job ID for further processing in local LINUX system • Optional: QV and length filtering of the corrected.fastq on the LINUX command line • Assemble corrected.fastq file via Celera® Assembler on the command line Exporting Data to Use for Celera® Assembler
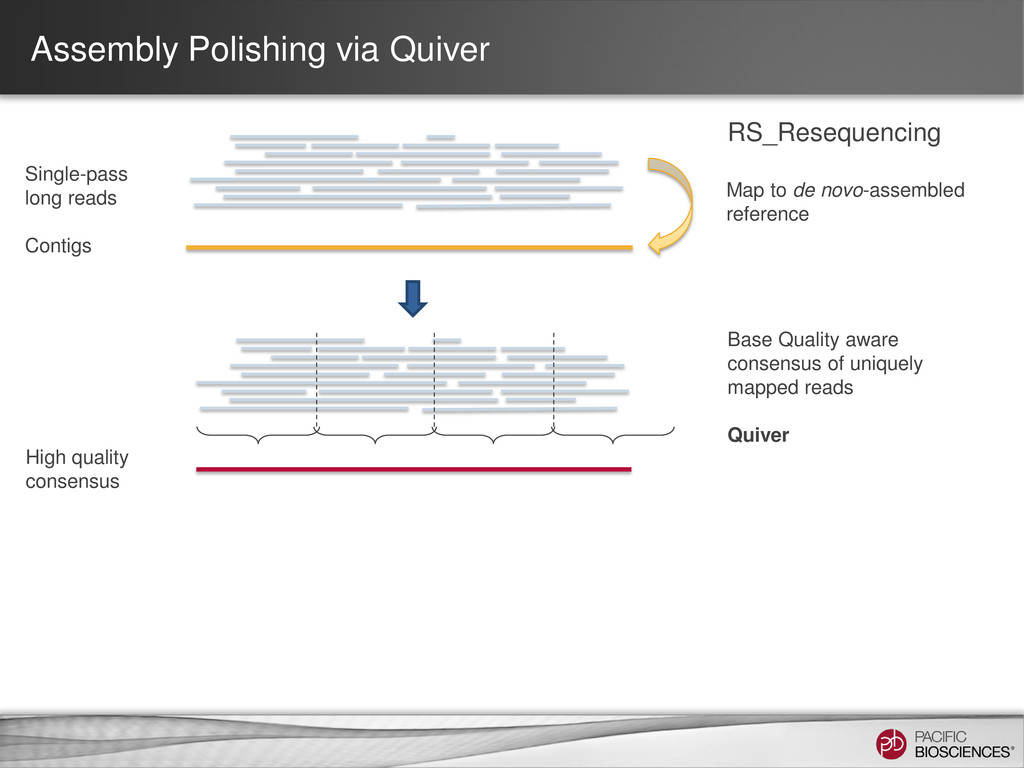
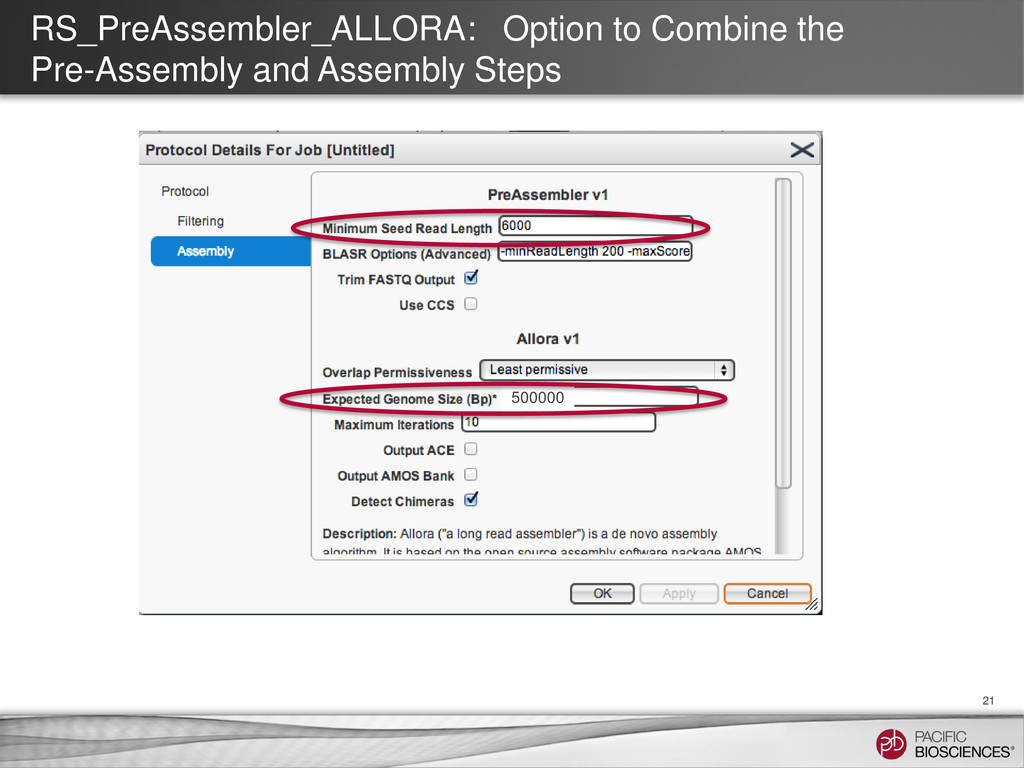
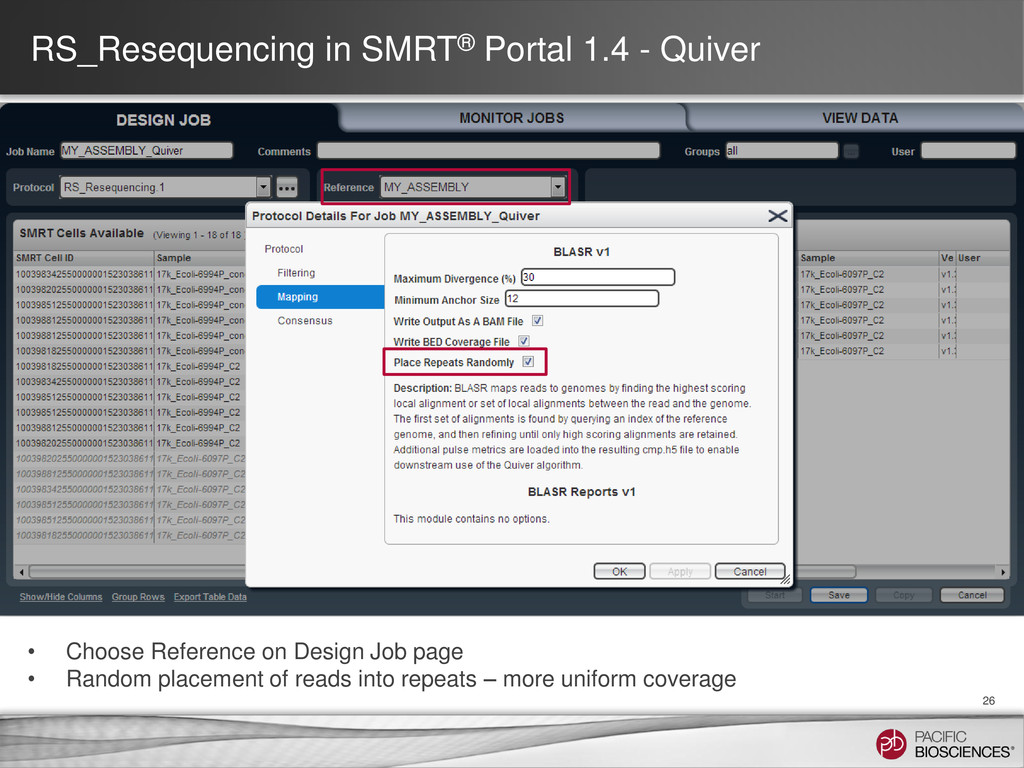
reads Tools: SMRT® Portal: RS_PreAssembler_ALLORA Command-line: RS_PreAssembler or P_PreAssembler Pre-Assembly Join reads into a near perfect assembly Tools: SMRT® Portal: RS_PreAssembler_ALLORA Command-line: Celera® Assembler Assembly Realign reads against the assembly for the highest final accuracy Tools: SMRT® Portal: RS_Resequencing Command-line RS_Resequencing or P_GenomicConsensus Assembly Polishing
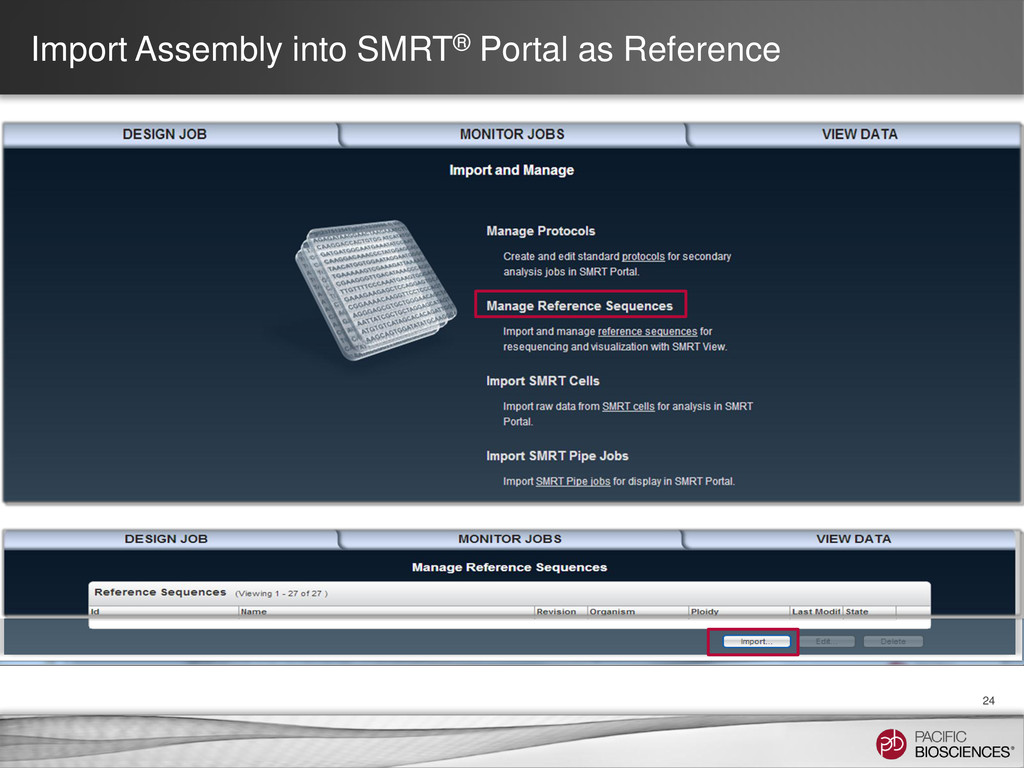
consider: • Scientist level SMRT Portal users can now delete their own single-use assembly references after finishing Quiver • Multiple fasta files can be combined into one reference via <SHIFT><Select> • Depositing a fasta file in the reference_dropbox requires write access to the directory
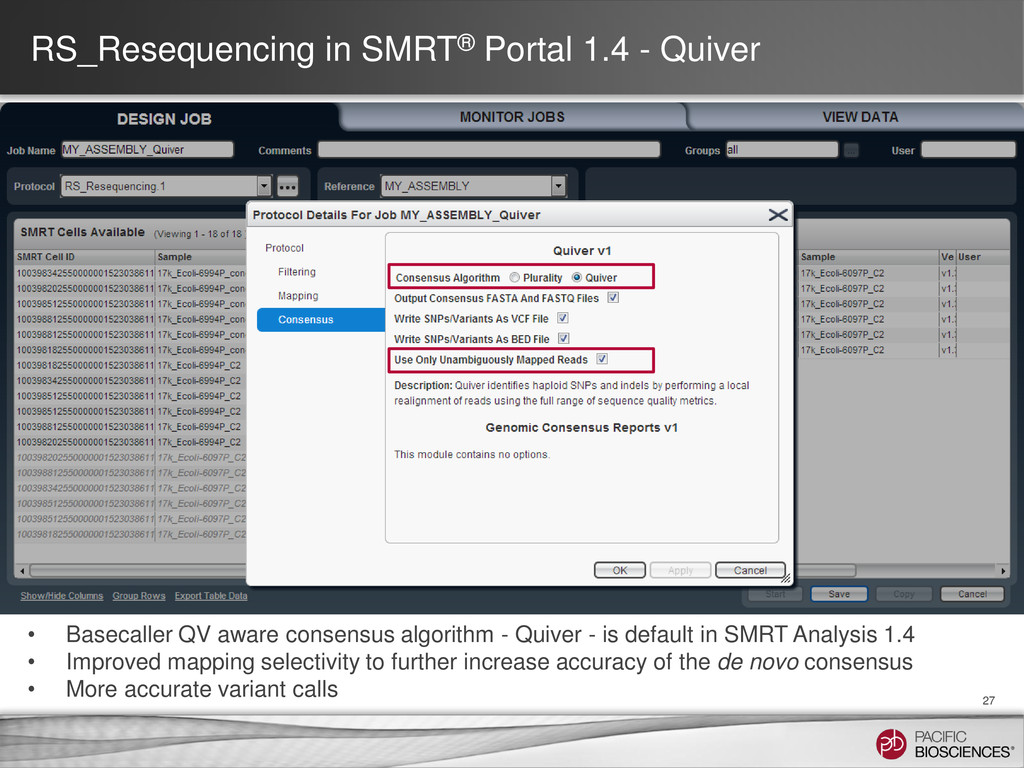
QV aware consensus algorithm - Quiver - is default in SMRT Analysis 1.4 • Improved mapping selectivity to further increase accuracy of the de novo consensus • More accurate variant calls
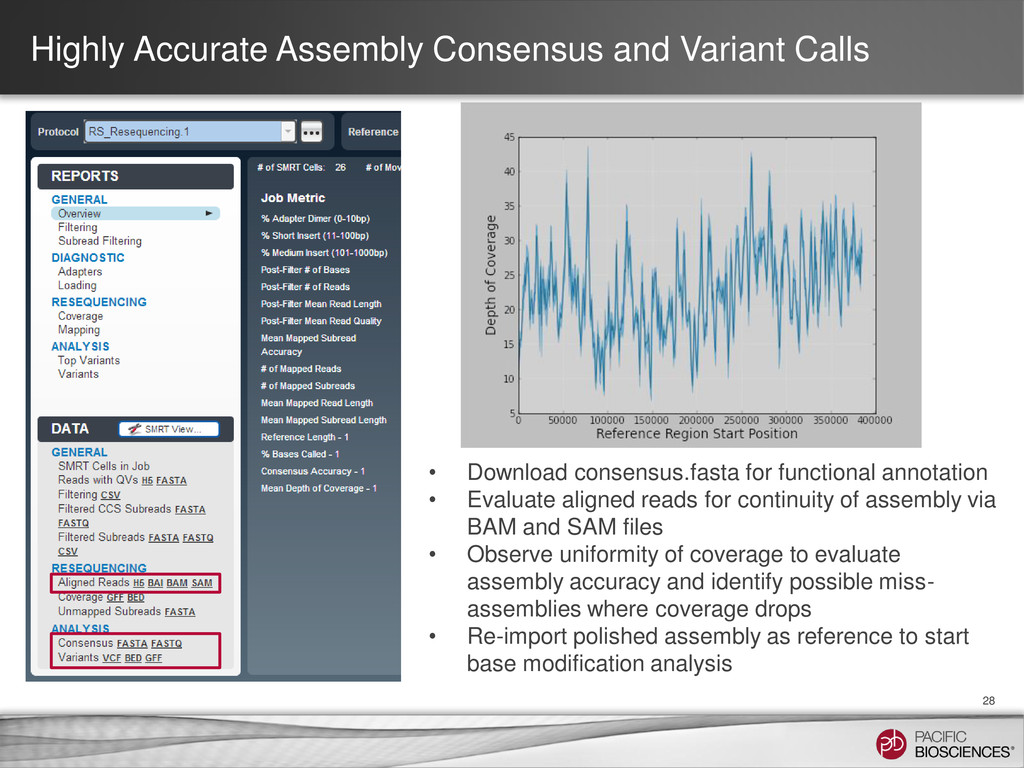
consensus.fasta for functional annotation • Evaluate aligned reads for continuity of assembly via BAM and SAM files • Observe uniformity of coverage to evaluate assembly accuracy and identify possible miss- assemblies where coverage drops • Re-import polished assembly as reference to start base modification analysis
P_PreAssembler with SMRT® Pipe on the command-line • Run Celera® Assembler on the command-line • Use Quiver option of P_GenomicConsensus to polish the assembly • For advanced users: – Additional tweaks to filtering and trimming may improve assembly – A beta release of HGAP on DevNet may generate even better assemblies (separate installation required) • More details here: https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/HGAP 29
Sequencing on the PacBio® RS and primary analysis Secondary Analysis Tertiary Analysis • XL-C2 chemistry • MagBead loading • Stage start • Movie Time: 1 x 120 min • Alternative movie times can be explored to optimize throughput • Do not overload; Loading titrations may be useful • 1.4 RS_Preassembler+Celera® Assembler SMRT® Analysis • Cov: 100 X • Use XL parameters, custom trimming as necessary • Recommend Quiver for assembly polishing to increase consensus accuracy • Base modification caveats • Limit DNA damage during sample extraction • 10 kb library protocol for long read library • Optional >10 kb protocol available through SampleNet • Good quality sample preparation is key!
has tested HGAP primarily on microbial-sized genomes. In principle, HGAP will work on genomes of 100 MB or larger, but this has not yet been tested, and manual fine-tuning will likely be necessary to achieve the best assembly. Q. What if customers have been using the DevNet implementation of HGAP? For advanced users who are comfortable installing beta software, the DevNet implementation (called the “reference implementation” or “beta”) is also available. • Advantages: potentially more scalable for larger genomes >500 MB. • Disadvantages: separate installation, command-line only, and may not be better in all cases. Q. What are the future plans for HGAP in SMRT® Analysis? In the upcoming release of SMRT Analysis 2.0, HGAP will be an integrated protocol in SMRT Portal, combining the Pre- Assembler with Celera® Assembler. Q. What about Celera® Assembler? Will CA implement PacBio long-read-only assembly in the future? A pre-release version of pacBioToCA can perform the preassembly step. More information can be found at http://sourceforge.net/apps/mediawiki/wgs-assembler/index.php?title=PacBioToCA. Celera Assembler can perform the assembly step. It’s still necessary to run resequencing with Quiver to polish the final assembly. We do not know when the Celera Assembler update officially will be released. We will evaluate including the update in a future version of SMRT Analysis. Q. Where can I get more information about HGAP? See pacbiodevnet.com for more details; in particular: https://github.com/PacificBiosciences/Bioinformatics-Training/wiki/HGAP 31
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}