Hypermobile Ehlers-Danlos syndrome & hypermobility spectrum disorders -A presentation.
Hypermobile Ehlers-Danlos syndrome & hypermobility spectrum disorders - A presentation.
I put this together for my own learning and to present to my professional peers.
most common genetic connective tissue disorder, but is commonly misdiagnosed (as are all HSD). Prevalence is unclear with a dearth of up-to-date research. It was estimated that hEDS affects 1 in 5,000 people globally (0.02% of population), but a study published this year suggests this number is at least 1 in 500 (0.2%). Comparative prevalence; familial hyperlipidaemia; 1 in 500, cystic fibrosis; 1 in 1250. Huntington's disease; 1 in 15,000. hEDS & HDS may be inherited, or may occur as a new mutation - either way they usually cause the body's collagen to be fragile and stretchy. As a result, typical features include increased skin elasticity and/or increased joint mobility. As collagen is present throughout the body, people with hEDS & HDSs experience a broad range of symptoms. These are complex syndromes affecting many systems of the body at once. Symptoms and severity vary considerably from person to person.
hEDS is diagnosed. There is no genetic test for this type, so diagnosis involves looking for joint hypermobility, signs of faulty connective tissue throughout the body (hernias, prolapses, skin features), a family history and musculo-skeletal problems (long-term pain, dislocations). There are many associated symptoms and disorders which don't form part of the formal criteria, and which do not directly result from joint hypermobility, for example; orthostatic tachycardia, digestive disorders, pelvic and bladder dysfunction and anxiety disorders. These are often more detrimental to quality of life than the joint symptoms. Joint hypermobility with its possible complications is now classified using the idea of a spectrum (next slide). At one end is simple hypermobility with no symptoms - not a disease but a trait, like height. At the other end of the spectrum is hEDS and in between falls a range of hypermobility-related conditions called hypermobility spectrum disorders (HSD).
can be just as symptomatic - more so, even - than someone with hEDS. Management advice for both hEDS and HSD is the same. THE 'HYPERMOBILITY SPECTRUM'.
of which are very rare. The gene mutations causing these conditions have been identified and can be tested for in all types, except the most common - hypermobile EDS. Hypermobile (hEDS) - most common. Kyphoscoliotic (kEDS). Arthrochalasia (aEDS). Dermatosparaxis (dEDS). Brittle cornea syndrome (BCS). The rest are extremely rare; Myopathic (mEDS). Periodontal (pEDS). Cardiac-valvular (cvEDS). Classical (cEDS) - rare. Vascular (vEDS) - rare. Classical-like (clEDS). Spondylodysplastic (spEDS). Musculocotractual (mcEDS).
hEDS. 80-99% of individuals will have the following; Acrocyanosis (persistent blue colour of hands, feet Arthralgia (joint pain). Elbow dislocation. Fatigue. Hip dislocation. Hyperextensible/hyperelastic skin. Joint hyperflexibility. Myalgia (muscle ache). Sleep disturbance. Vertigo. Wormian bones (extra bones with cranial sutures). or parts of face). (hEDS). 30-79% will have the following; Arrhythmia. Constipation. Decreased nerve conduction velocity. Depression. Malabsorption. Migraine. Nausea and vomiting. Osteoarthritis. Pes planus (flat feet). Soft skin. Thin skin.
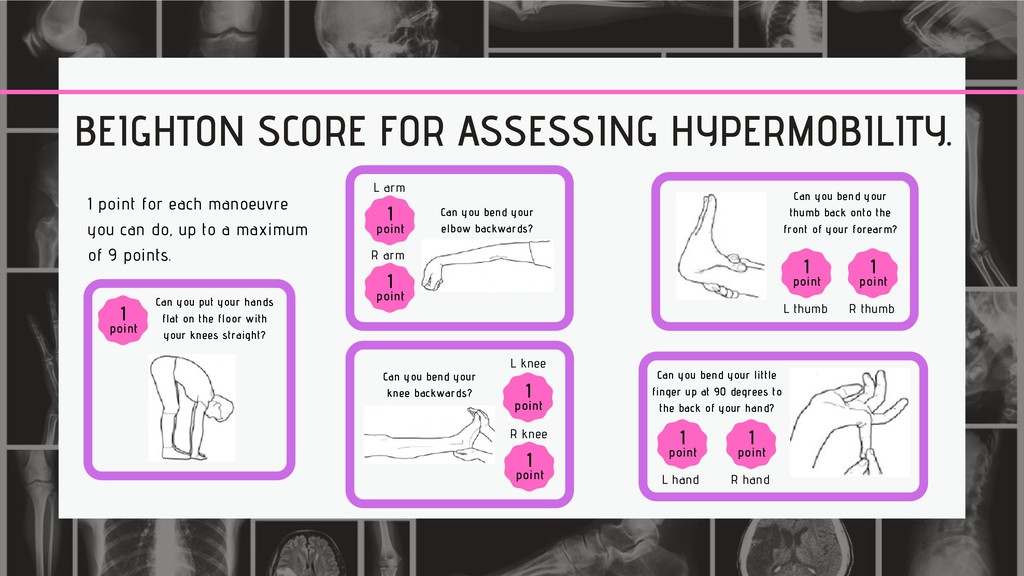
you can do, up to a maximum of 9 points. Can you put your hands flat on the floor with your knees straight? 1 point 1 point L arm R arm 1 point Can you bend your elbow backwards? Can you bend your knee backwards? L knee 1 point R knee 1 point Can you bend your thumb back onto the front of your forearm? R thumb 1 point L thumb 1 point L hand 1 point R hand 1 point Can you bend your little finger up at 90 degrees to the back of your hand?
there is no molecular or genetic cause yet identified, so there is no test available. There is a clinical spectrum ranging from asymptomatic joint hypermobility, through "non-syndromic" hypermobility with secondary manifestations, to hEDS. A diagnosis of hEDS should be assigned only in those who meet all of the criteria, which should help research efforts to identify the underlying genetic cause(s), which in turn may help clinical management. As this is a clinical diagnosis, it is important to be relatively confident that the diagnosis is not instead one of the many other disorders of connective tissue, for example; Marfan Syndrome, Osteogenesis imperfecta, Sjogren's syndrome and many others. The international diagnostic criteria for hEDS is shown on the right. (hEDS).
form of EDS. Long-term prognosis is generally poor and is associated with reduced life expectancy. Estimated to affect between 1 in 50,000 and 1 in 200,000. Amongst those diagnosed as the result of a complication, 25% have a significant medical complication by age 20 and >80% by age 40. Median life span is 48 years. Some people are diagnosed on the basis of subtle signs in their physical appearance together with medical history; Fragile tissues (including arteries, muscles and internal organs) prone to rupture. Thin, translucent skin. Characteristic facial appearance (thin lips, small chin, thin nose, large eyes). Acrogeria (premature aging of hands and feet). Hypermobility of small joints. Early onset varicose veins. Pneumothorax. Easy bruising. Joint dislocations and subluxations. Congenital dislocation of the hips. Congenital clubfoot. Receding gums.
a genetic test. Sometimes a skin biopsy is required for confirmation. Individuals may not experience any significant problems until later in life. Others will have had signs of vEDS from a young age. Day to day, many individuals with vEDS are physically fit and well, however there is a risk of problems due to fragile blood vessels and hollow organs which could rupture. These are unpredictable and usually result in emergency situations. As a result it is important to discern if a patient has this diagnosis to aid in timely management. Activities that increase risk; Strenuous contact sports. Sprinting / activities involving sudden acceleration. Tasks involving pushing or lifting heavy objects. Competitive exercise performed to the point of exhaustion. Playing a brass instrument causing an increased in pressure in the lungs. Activities to be encouraged include regular aerobic exercise performed in moderation, with examples as follows; Swimming, cycling, walking, hiking, jogging.
be seen annually in a specialist cardiac clinic. This ensures access to up to date evidence-based care. Management priorities include control of blood pressure, and scans to exclude development of any aneurysms. General advice is to avoid surgery where possible. Fragility of tissues and blood vessels significantly increases risk of serious complications. Pregnancy puts the cardiovascular system under extreme pressure in all individuals and there are additional risks for women with vascular EDS in pregnancy. It is important that this is communicated early in the pregnancy. A planned caesarean delivery in a hospital with specialist vascular surgery may be suggested. Family history of vEDS with documented causative variant in COL3A1; Arterial rupture at a young age; Spontaneous sigmoid colon perforation in the absence of known diverticular disease or other bowel pathology; Uterine rupture during the 3rd trimester in the absence of previous C-section or severe peri-partum tears; Carotid-cavernous sinus fistula (CCSF) formation in the absence of trauma. Major diagnostic criteria; 1. 2. 3. 4. 5. There are 12 minor criteria - Minimal clinical standards suggesting vEDS are a family history of the disorder, arterial rupture or dissection in individuals less than 40 years old, unexplained sigmoid colon rupture, or spontaneous pneumothorax in the presence of other features consistent with vEDS. Testing for vEDS should also be considered in the presence of a combination of the other minor criteria. Final diagnosis is made by molecular testing.
hypermobility, very stretchy and fragile skin which leads to significant bruising and widened, sunken (atrophic) scars. As with vEDS, it is often possible to make a diagnosis of cEDS from physical examination and medical history. Clinical features to look for include; Fragile skin prone to splitting with minimal trauma. This leads to significant scarring usually starting in childhood. Common sites are knees, elbows, shins, forehead and chin. Scars tend to be wide with a thin appearance often described as "like tissue paper" (surgical scars tend to heal normally). Stretchy skin, often very stretchy! Joint hypermobility , which may cause them to slip out of position resulting in dislocations or subluxations and may be associated with chronic joint pain. Easy bruising, which may lead to permanent discolouration and is often visible on the shins. Fragile and extensible tissues can also result in hernias, prolapse and cervical insufficiency.
living with cEDS is the fragility of their skin. It is therefore helpful to try to protect the skin against injury and ensure wounds are well stitched to help reduce scarring. High risk activities to avoid; Contact sports such as rugby, ice hockey. boxing and martial arts. Football and basketball are discouraged despite not being true contact sports. Playing at a competitive level is discouraged. If joint dislocations are a problem, then certain activities such as trampolining is also discouraged. Activities to be encouraged; General health and regular gentle exercise such as badminton, squash, table tennis, bowling, walking, swimming. Fatigue and joint pain can be features of cEDS so this helps reduce these. Adults may benefit from pilates as this builds core strength and protects joints. Physiotherapy may be helpful for significant joint hypermobility. Occupational therapy may be able to make recommendations for appropriate aids to assist with activities of daily living where required, and also advice on pacing of activities to avoid 'boom and bust' phenomenon and extreme fatigue.
in cEDS. It is generally accepted that trans-thoracic echocardiography (TTE) should be carried out regularly due to an association with valve prolapse, particularly mitral. Higher risk of tearing in pregnancy and also a higher risk of early rupture of membranes and early delivery if either parent has cEDS. Other features of cEDS; Piezogenic pedal papules - fat lumps that are visible around the heel of the foot (below). Molluscoid pseudotumours - fleshy lesions over elbows and knees associated with scars (bottom). Subcutaneous spheroids - small hard nodules that are movable under the skin, due to fat lobules that have lost blood supply and calcified. People with cEDS do not get stretch marks, even women who have had multiple pregnancies. Skin hyperextensibility and atrophic scarring; Generalised joint hypermobility. Major diagnostic criteria; 1. 2. There are 9 minor criteria. Minimal clinical standards suggesting cEDS are major criterion 1 plus either major criterion 2 or at lest 3 minor criteria. Final diagnosis is made by molecular testing.
in the medical literature is based on a very small number of cases. Most healthcare professionals will never see a patient with any of these conditions in their whole career. ARTHROCHALASIA EDS (aEDS). DERMATOSPARAXIS EDS (dEDS). BRITTLE CORNEA SYNDROME (BCS). There are 5 minor criteria. Minimal clinical standards suggesting aEDS are major criterion 1 plus either major criterion 2 or 3, with at least 2 minor criteria.. Final diagnosis is made by genetic testing. Congenital bilateral hip dislocation; Severe generalised joint hypermobility with multiple dislocations/subluxations; Skin hyperextensiblity. Major criteria; 1. 2. 3. There are 9 major criteria and 11 minor criteria. Minimal clinical standards suggesting dEDS are 2 major criteria of extreme skin fragility and characteristic cranio-facial features, plus either 1 other major criterion or 3 minor criteria. Final diagnosis is made by genetic testing. (Characteristic cranio-facial features include puffy eyelids, blue sclerae, epicanthal folds (skin of the upper eyelid that covers the inner corner of the eye), down-slanting palpebral fissures (corners of the eyes that point downward) and micrognathia (undersized jaw). There are 10 minor criteria. Minimal clinical standards suggesting kEDS are major criteria 1 and 2 plus either major criterion 3 or 3 or more minor criteria.. Final diagnosis is made by genetic testing, although some kEDS patients can be confirmed via a urine sample using high performance liquid chromotography.. Congenital muscle hypotonia; Congenital or early onset kyphoscoliosis (progressive or non-progressive); Generalised joint hypermobility with dislocations/subluxations (shoulders/hips/knees in particular. Major criteria; 1. 2. 3. Thin cornea with or without rupture (central corneal thickness often >400um); Early onset progressive keratoconus (cone shaped cornea); Early onset progressive keratoglobus (globular shaped cornea); Blue sclerae. Major criteria; 1. 2. 3. 4. There are 14 minor criteria. Minimal clinical standards suggesting BCS are major criterion 1 plus 1 other major criterion, or 3 or more minor criteria.. Final diagnosis is made by genetic testing.
texture and absence of atrophic scarring; Generalised joint hypermobility with or without recurrent dislocations (often shoulder or ankle); Easily bruised skin or spontaneous ecchymoses (skin discolorations due to bleeding under the skin). Major criteria; 1. 2. 3. There are 7 minor criteria. Minimal clinical standards suggesting clEDS are all 3 major criteria plus a family history compatible with autosomal recessive transmission. Final diagnosis is made by genetic testing. CLASSICAL-LIKE EDS (clEDS). Information in the medical literature is based on a very small number of cases. Most healthcare professionals will never see a patient with any of these conditions in their whole career. SPONDYLODYSPLASTIC EDS (spEDS). MUSCULOCONTRACTUAL EDS (mcEDS). MYOPATHIC EDS (mEDS). (only 11 cases - all within 6 families - reported WORLDWIDE). Short stature (progressive in childhood); Muscle hypotonia (ranging from severe congenital to mild late-onset); Bowing of limbs; Major criteria; 1. 2. 3. There are 5 minor criteria. Minimal clinical standards suggesting spEDS are major criterion 1 and 2, plus characteristic radiographic abnormalities and at least 3 minor criteria. Final diagnosis is made by molecular testing. Congenital multiple contractures, characteristically adduction-flexion contractures and/or talipes equinovarus (clubfoot); Characteristic cranio-facial features, which are evident at birth or in early infancy; Characteristic cutaneous features including skin hyperextensibility, easy bruising, skin fragility with atrophic scar, increased palmar wrinkling. Major criteria; 1. 2. 3. There are 15 minor criteria. Minimal clinical standards suggesting mcEDS are major criteria 1 and 2 at birth or early childhood, or major criteria 1 and 3 in adolescence or adulthood. Final diagnosis is made by genetic testing. Congenital muscle hypotonia and/or muscle atrophy, that improves with age; Proximal joint contractures (knee/hip/elbow); Hypermobility of distal joints. Major criteria; 1. 2. 3. There are 4 minor criteria. Minimal clinical standards suggesting mEDS is major criterion 1 plus either 1 other major criterion, or 3 minor criteria. Final diagnosis is made by molecular testing.
based on a very small number of cases. Most healthcare professionals will never see a patient with any of these conditions in their whole career. PERIODONTAL EDS (pEDS). Severe and intractable periodontitis of early onset (childhood or adolescence); Lack of attached gingiva; Pretibial plaques; Family history of a first-degree relative who meets clinical criteria. Major criteria; 1. 2. 3. 4. There are 8 minor criteria. Minimal clinical standards suggesting pEDS are major criteria 1 or 2, plus at least 2 other major criteria and 1 minor criteria. Final diagnosis is made by molecular testing. CARDIAC-VALVULAR EDS (cvEDS). Severe progressive cardiac-valvular problems (aortic valve, mitral valve); Skin involvement: skin hyperextensibility, atrophic scars, thin skin, easy bruising; Joint hypermobility (generalised or restricted to small joints). Major criteria; 1. 2. 3. There are 4 minor criteria. Minimal clinical standards suggesting cvEDS are major criterion 1, plus a family history compatible with autosomal recessive transmission and either 1 other major criterion or 2 or more minor criteria. Final diagnosis is made by genetic testing.
of the mainstays of managing the conditions of hEDS/HSDs. There should be both a subjective assessment and an objective assessment, to allow for a tailored physiotherapy regime to best meet the needs of the individual. Physiotherapy. Chronic pain in the Ehlers-Danlos syndromes, especially hEDS, is common and can be severe. It may be widespread or localised. Headaches and gastrointestinal discomfort as well as joint, muscular and nerve pain can occur. Management includes physiotherapy, medications and supports such as splints and braces. Pain management. There is an overlap in the symptoms of hEDS and ME/chronic fatigue syndrome - but they are clinically distinct. There is no medication to alleviate tiredness, but certain drugs are effective for problems that contribute to fatigue. Physiotherapy, prevention of deconditiong and good nutrition are recommneded. Fatigue. Orthopaedic surgery in the Ehlers-Danlos syndromes is contraversial and how much of a role it should play in their management is unclear. Conservative managment is preferrable but if this fails, joint stabilisation and nerve decompression procedures can provide relief. Surgery. hEDS/HSD cannot be 'cured', but many people learn over time to control it and live full and active lives. Day-to- day management of most types of EDS is supportive and based around appropriate exercise, physiotherapy and pacing of activities. In addition, referrals should be sought for any associated conditions. This may mean referral to specialist services such as neurology, gastroenterology, podiatry or pain management.
ED syndromes, particularly with hEDS and HSD. In many of these cases, whilst there is a demonstrable association, it is not scientifically possible to prove that one causes the other. Brain and spine; Migraine. Early disc degeneration. Chiari 1 malformation. Craniocervical instability. Motor delay. Curvature of the spine. MSK pain. Weakness. Fatigue. Mobility impairment. Joints; Curvature and twisting of the spine. Early osteoparosis. Osteoarthritis. Dislocations. Scoliosis and kyphosis. Instability of the craniocervical junction. Skin; Fragility. Stretchiness. Abnormal scarring. Stretch marks. Easy bruising. Slow wound healing. Immune system; Mast cell activation. Excess histamine production.
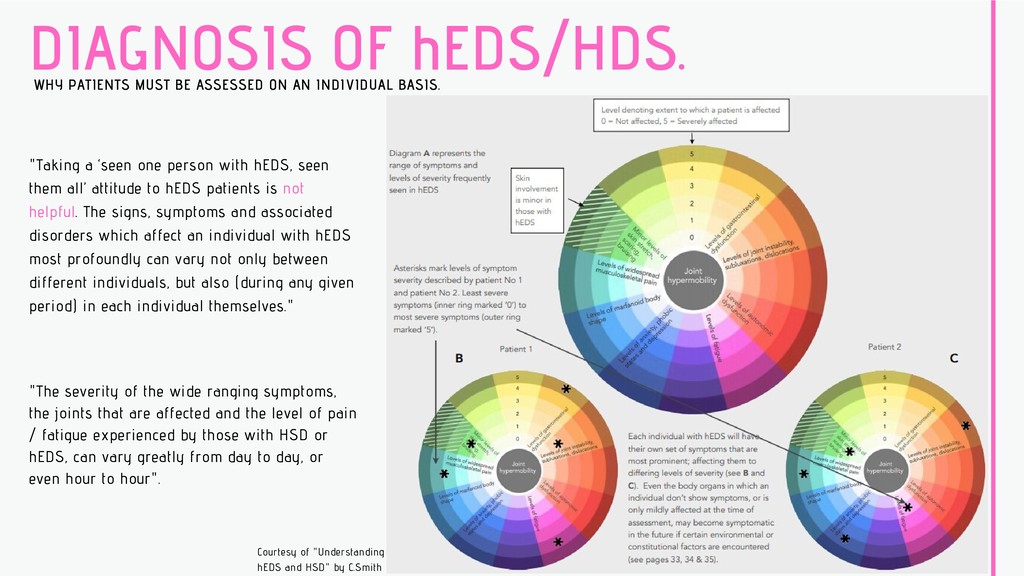
INDIVIDUAL BASIS. "Taking a ‘seen one person with hEDS, seen them all’ attitude to hEDS patients is not helpful. The signs, symptoms and associated disorders which affect an individual with hEDS most profoundly can vary not only between different individuals, but also (during any given period) in each individual themselves." "The severity of the wide ranging symptoms, the joints that are affected and the level of pain / fatigue experienced by those with HSD or hEDS, can vary greatly from day to day, or even hour to hour". Courtesy of "Understanding hEDS and HSD" by C.Smith
- especially in vascular EDS. Severe progressive problems of the aortic and mitral valves in cardiac-valvular EDS. In hEDS, you're likely to see a (mild) dilation of the aortic root - this is 'unlikely to progress'). Association with postural orthostatic tachycardia syndrome (PoTS). Whether there is an increased frequency of mitral valve prolapse in hEDS is contraversial. LOW THRESHOLD FOR CT-AORTAGRAM IN THE ACUTE SETTING FOR ALL FORMS OF EDS AGGRESSIVE CONTROL OF BLOOD PRESSURE MANY WILL HAVE AN INDICATION FOR SURVEILLANCE ECHOCARDIOGRAMS SEEK EXPERT OPINION WHERE A DIAGNOSIS IS SUSPECTED (SHORT TERM) (SHORT-MEDIUM TERM) (LONG TERM) (MEDIUM-LONG TERM) HIGHEST RISK OF THORACIC AORTIC DISSECTION IN VASCULAR EDS, FOLLOWED BY HYPERMOBILE AND KYPHOSCOLIOTIC EDS, BUT THRESHOLD FOR SUSPICION SHOULD BE LOW IN ALL hEDS/HSD. (PREVIOUS SLIDE)
long-standing joint pain and chronic fatigue syndrome to a primary care setting w/ new onset (R) shoulder pain for around 1 month, w/ a grinding quality and sensation of joint "displacement" when leaning on her right side. On review she was found to have chronic headache and joint pain, "sensitive skin" and general itchiness. PMH; MSK - temperomandibular joint (TMJ) disorder , chronic joint and muscle pain, lower back pain, and repetitive motion injuries. Her pain began around age 12. Lifelong history of injuries to joints, including frequent sprained ankles, which happened seemingly with no inciting event. Easy bruising. CV - diagnosis of orthostatic hypotension in her youth, which had never been medicated and of which she displayed ongoing symptoms. GI - irritable bowel syndrome and frequent nausea. She was being treated for Vitamin D and Bi2 deficiency. She had recently developed various chemical sensitivities, but no medication allergies. FH; Daughter under investigation for PoTS. (a typical case of un-diagnosed hEDS);
extremities, w/ pain to the rotator cuff. Patient scored 5/9 on Beighton scale criteria; bilateral 5th digit passively extended to 90 degrees; the (L) thumb was opposable to the forearm; bilateral elbow extension past 10 degrees, She was unable to palm the floor but said she would have been able to when younger prior to weight gain. Skin - soft, velvety skin w/ normal extensibility. Upon drawing a tongue depressor across her forearm, an erythematous wheal appeared in under 30 seconds. Patient was given a diagnosis of hypermobile EDS based on the findings of her physical examination; in particular the Beighton score, together w/ her history of joint pain and injury and TMJ disorder support hEDS. Velvety, doughy skin is a major indication of the diagnosis as well. Autonomic dysfunction (postural hypotension and IBS are commonly associated disorders. Referred into cardiology for TTE to rule out aortic root dilation, 'Max-fax' TMJ pain control and physiotherapy and rehabilitation for joint pain management. Very commonly mis-diagnosed/un-diagnosed. It seems obvious in retrospect; consider the full systemic picture. (a typical case of un-diagnosed hEDS); Learning point
retrosternal chest pain at rest. ECG; ST segment elevation v3-v4. PMH; Genetically confirmed Ehler-Danlos type IV (reclassified as vEDS since 2017) at age 24, after presenting with ruptured (L) common iliac artery aneurysm. Ruptured (L) renal artery dissection at age 26. CT at the time showed aneurysms of the celiac, (R) renal, superior mesenteric, (L) vertebral, (L) carotid, hepatic and (R) common iliac arteries. O/E; Notable for a tall, slim man w/ a diminutive chin, minimal earlobes and absence of hypermobile joints. (there's more to EDS than just thoracic aortic dissection);
aortic dissection); Coronary angiography demonstrated isolated proximal LAD involvement w/ coronary artery dissection extending to the mid segment. Patient transferred to cadiothoracic centre due to ongoing chest pain and haemodynamic instability, where percutaneous intervention was opted against due to extent of dissection and tissue fragility - therefore underwent saphenous vein bypass graft to the LAD. Suffered post-operative VF arrest on the CICU which responded to a single DC shock w/ ROSC. Shortly after developed hypotension and signs of cardiac tamponade. Chest opened at the bedside and active bleeding from the proximal anastomosis of the vein graft was controlled w/ digital compression. Taken back to theatre where an aortic rupture medial to the proximal vein graft anastamosis was repaired on cardio-pulmonary bypass with a pericardial patch and the vein graft was resewn to this patch. Discharged home 5/7 days later and well at 6/52 F/U. Higher risk of surgical complications. Always think dissection; but not just aortic! Learning point Learning point
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}