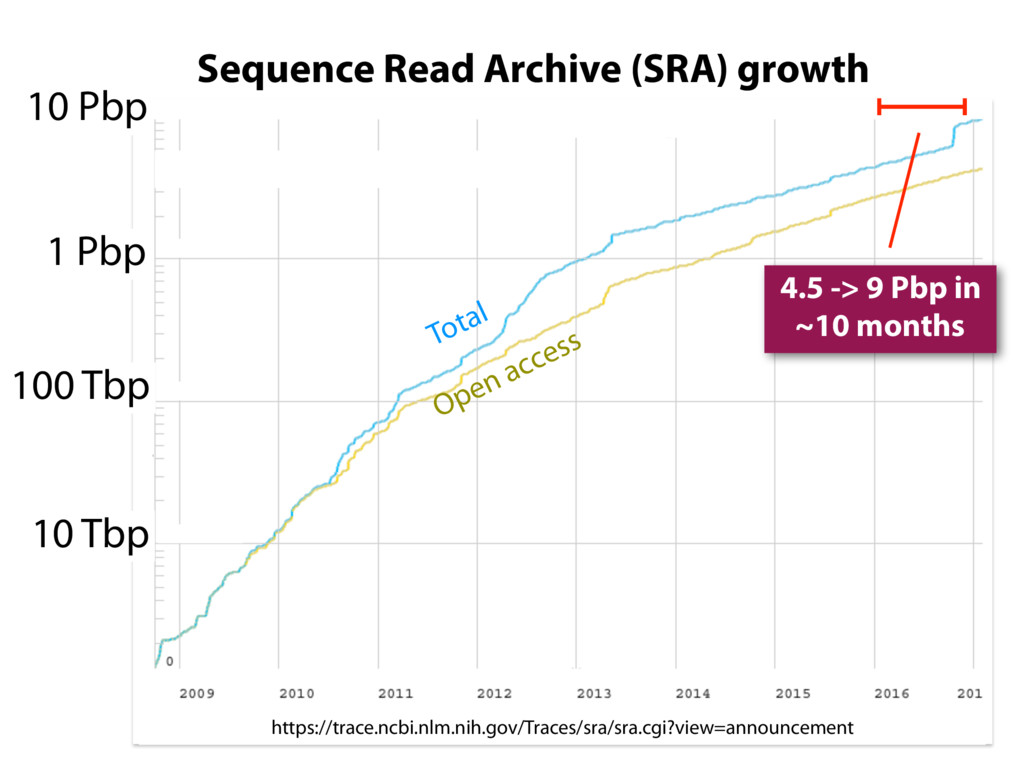
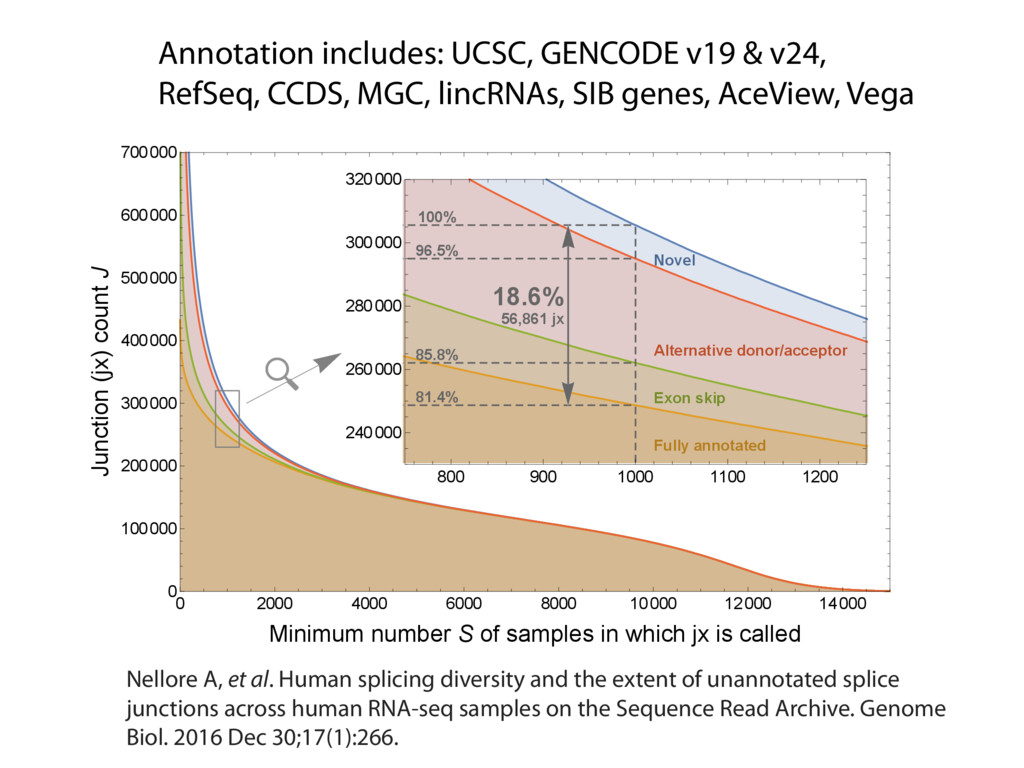
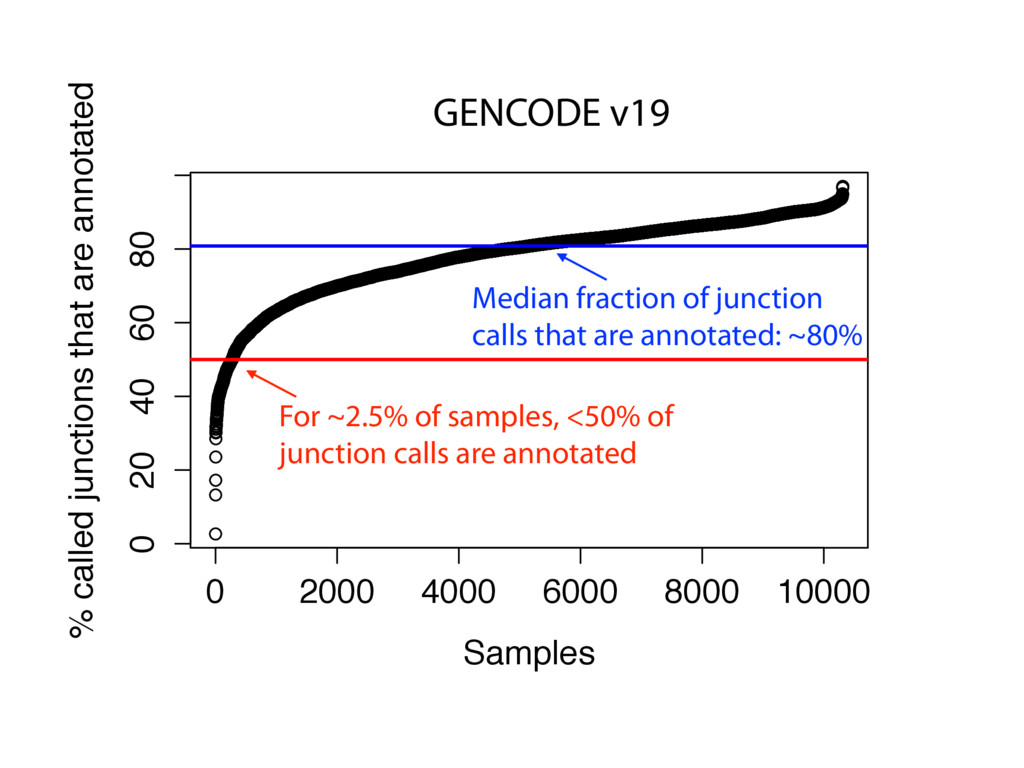
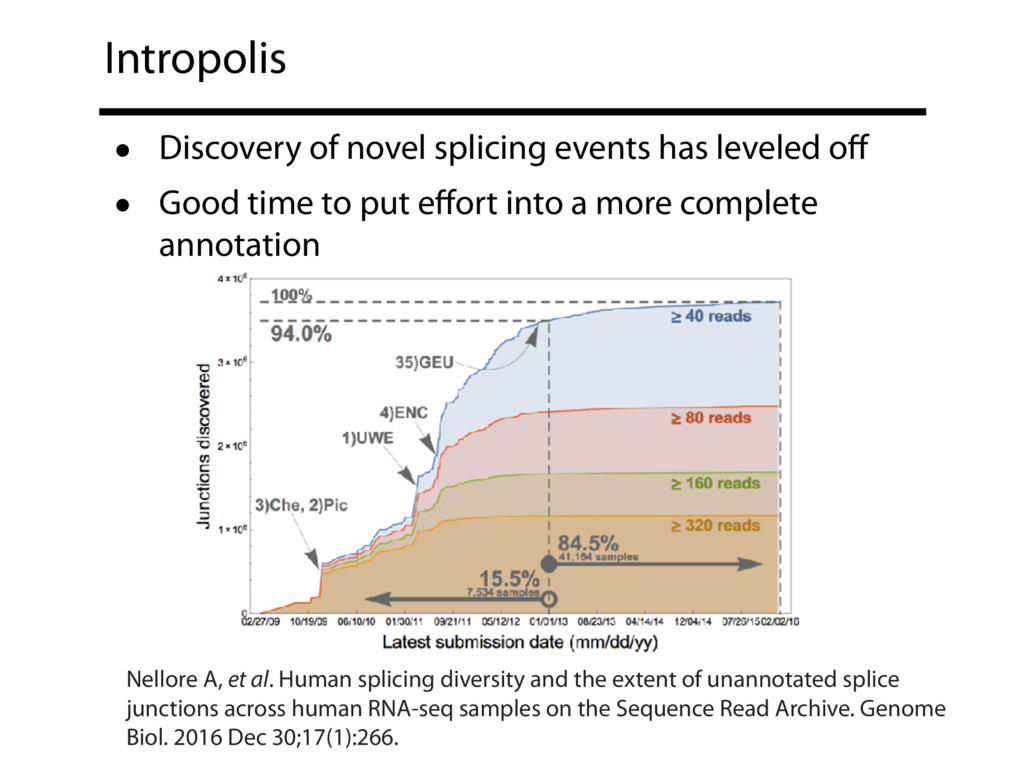
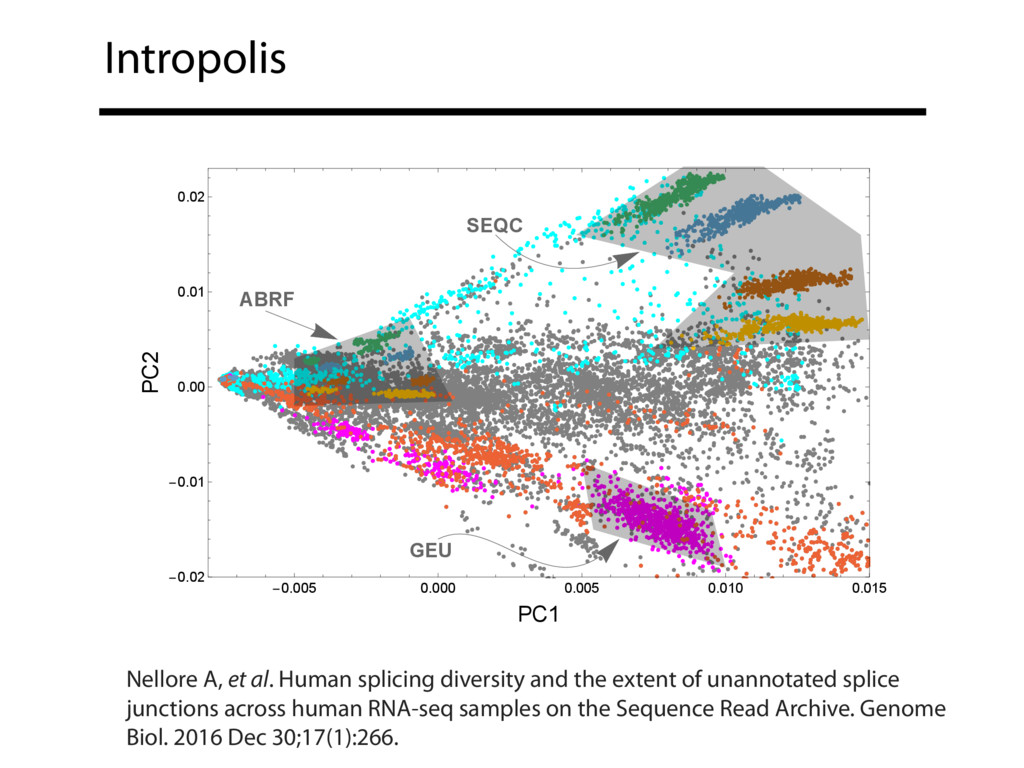
The Sequence Read Archive contains data for over 450K RNA-seq samples, including over 140K from human samples. Large-scale projects like GTEx and ICGC are generating RNA-seq data on many thousands of samples. Such huge and carefully designed datasets are valuable, but unwieldy for typical researchers, especially when access to computational resources is limited.
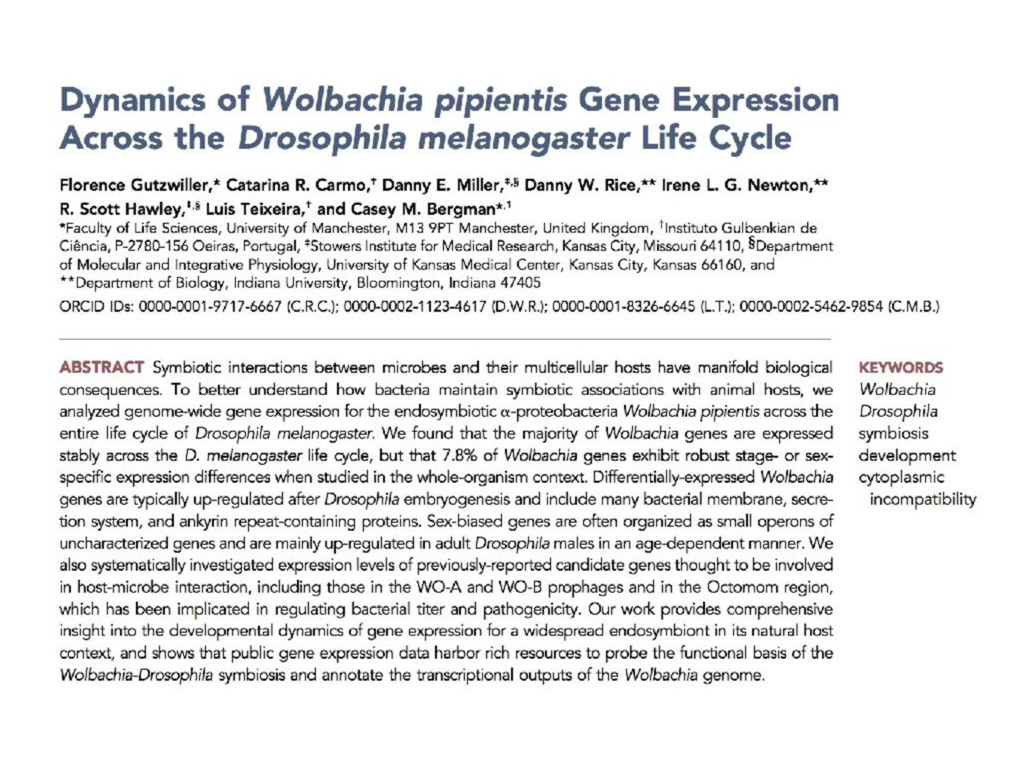
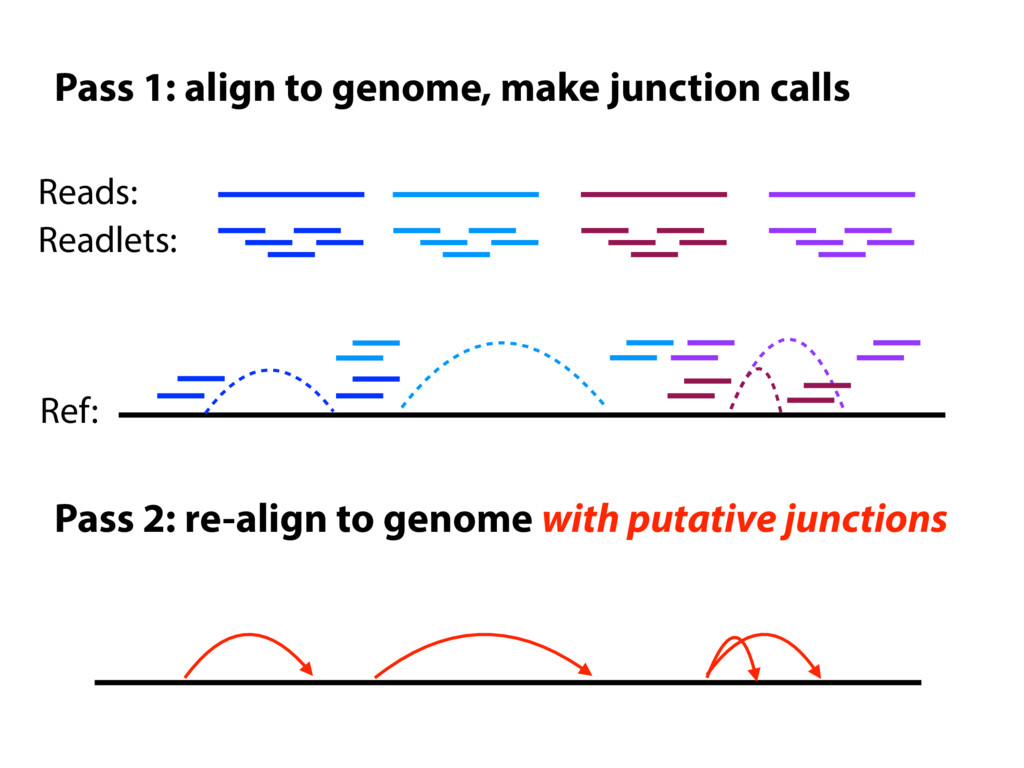
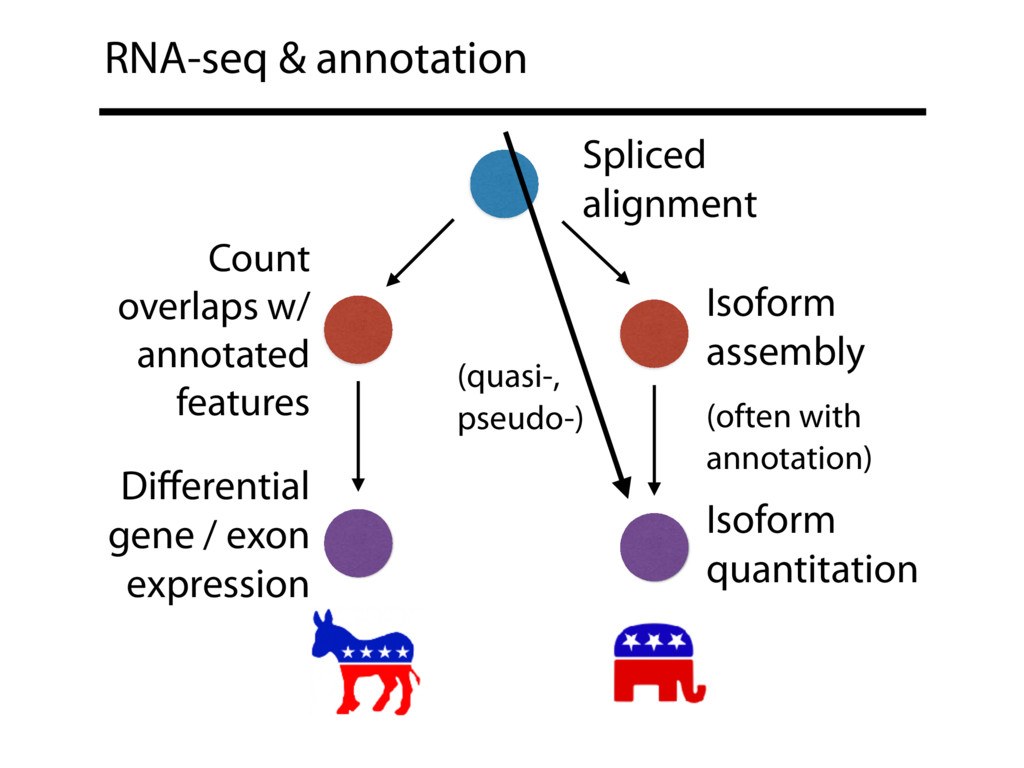
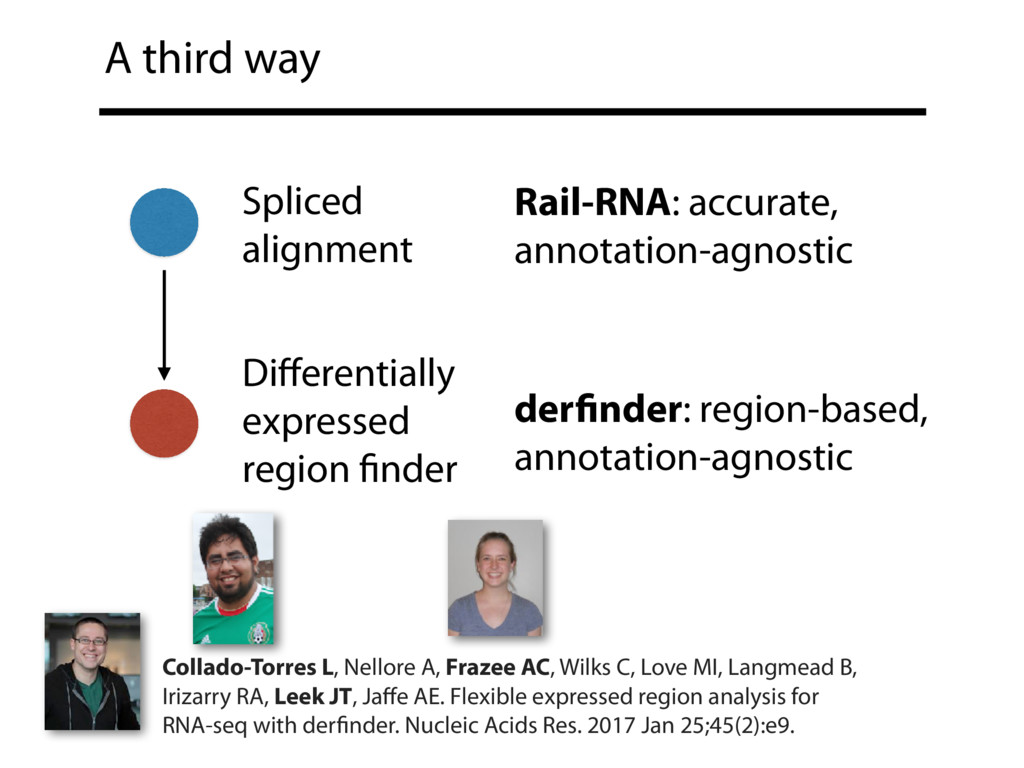
I will describe work toward the goal of making it easy for biological researchers to use the archived RNA-seq data available today. I will highlight the Rail-RNA software (http://rail.bio), its dbGaP-protected version (http://docs.rail.bio/dbgap/), as well as the recount (https://jhubiostatistics.shinyapps.io/recount/) and Snaptron (http://snaptron.cs.jhu.edu) resources. The Rail-RNA software uses the Amazon Web Services commercial cloud to analyze many samples at once. We used Rail-RNA to study tens of thousands of public RNA-seq accessions, yielding new insights about the completeness of existing gene annotations and about how our knowledge of human splicing diversity has evolved over time. I will demonstrate how the recount resource can be used to answer questions about expression and differential expression across 10,000s of RNA-seq samples, and how the Snaptron API can be used to rapidly answer sophisticated queries against the splicing patterns in recount.
![Ben Langmead Assistant Professor, JHU Computer Science [email protected], langmead-lab.org, @BenLangmead](https://files.speakerdeck.com/presentations/40ad19b6e31e4413854dca05a4af59d4/slide_0.jpg){kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
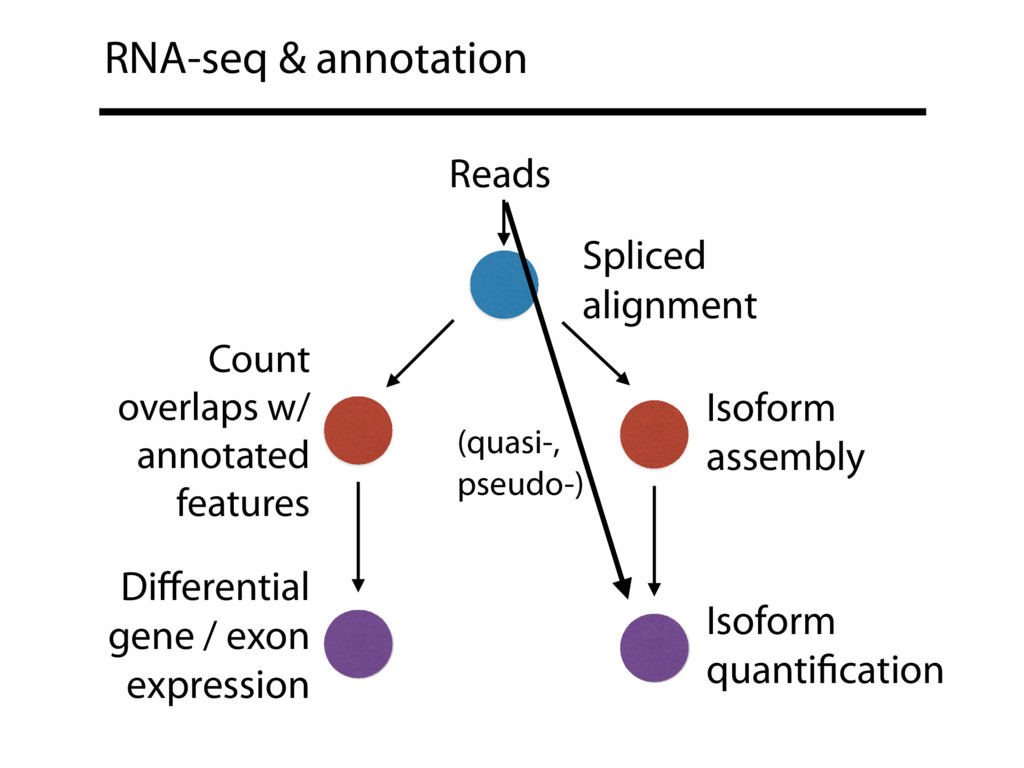{kind=link}
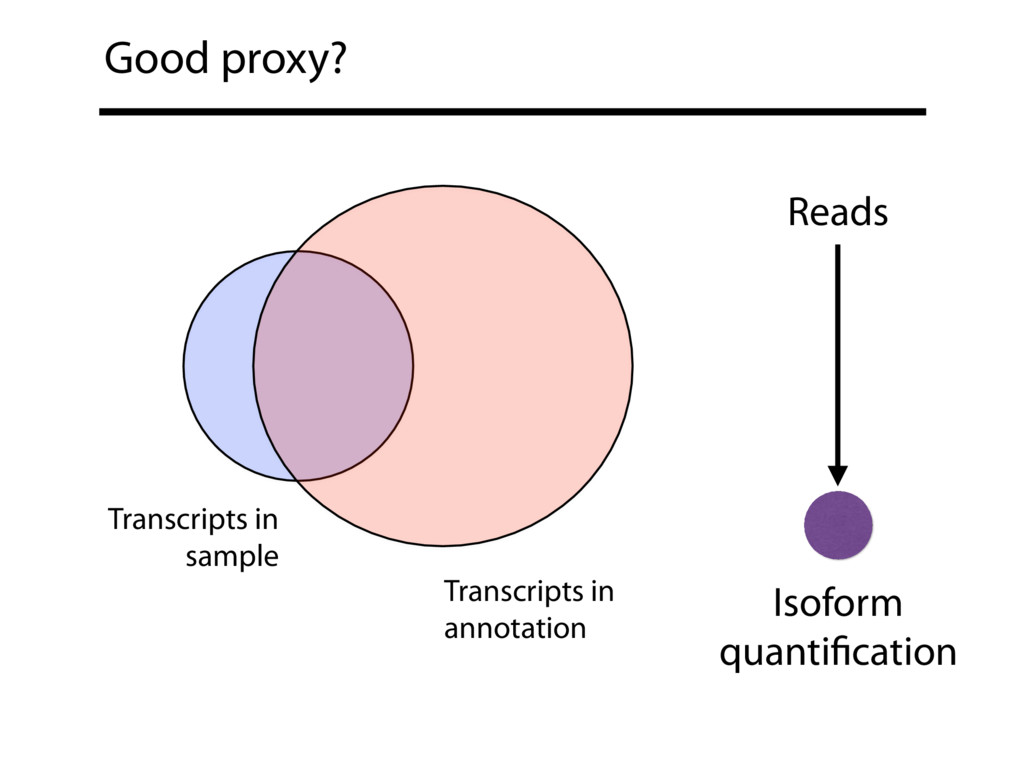{kind=link}
{kind=link}
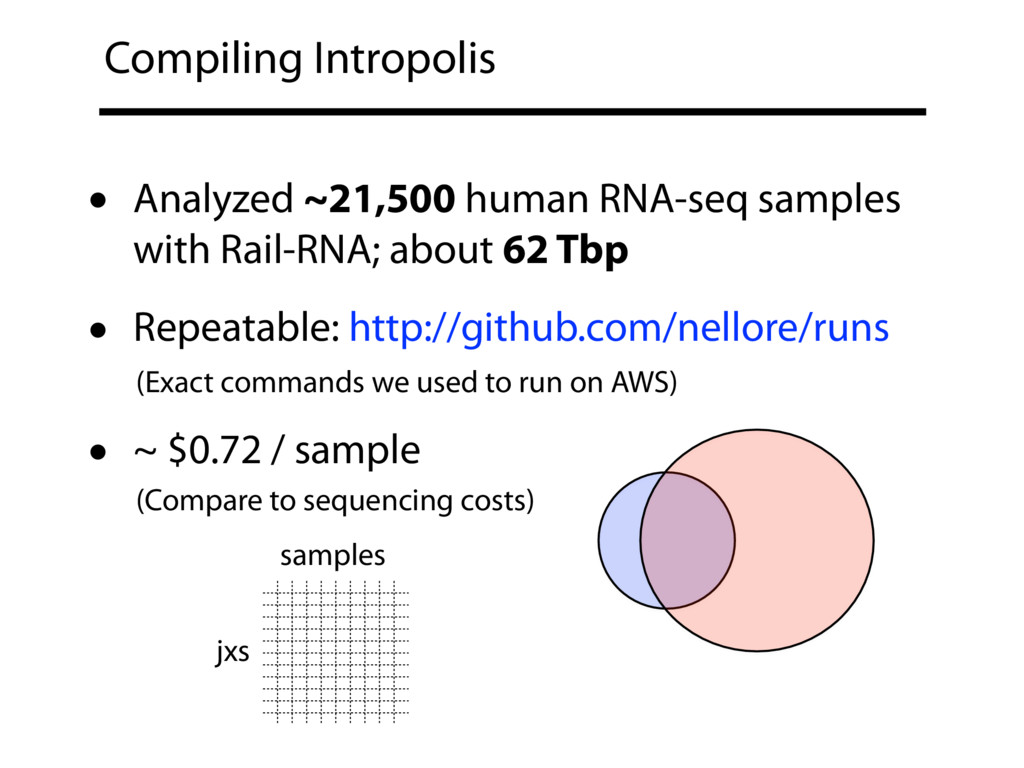{kind=link}
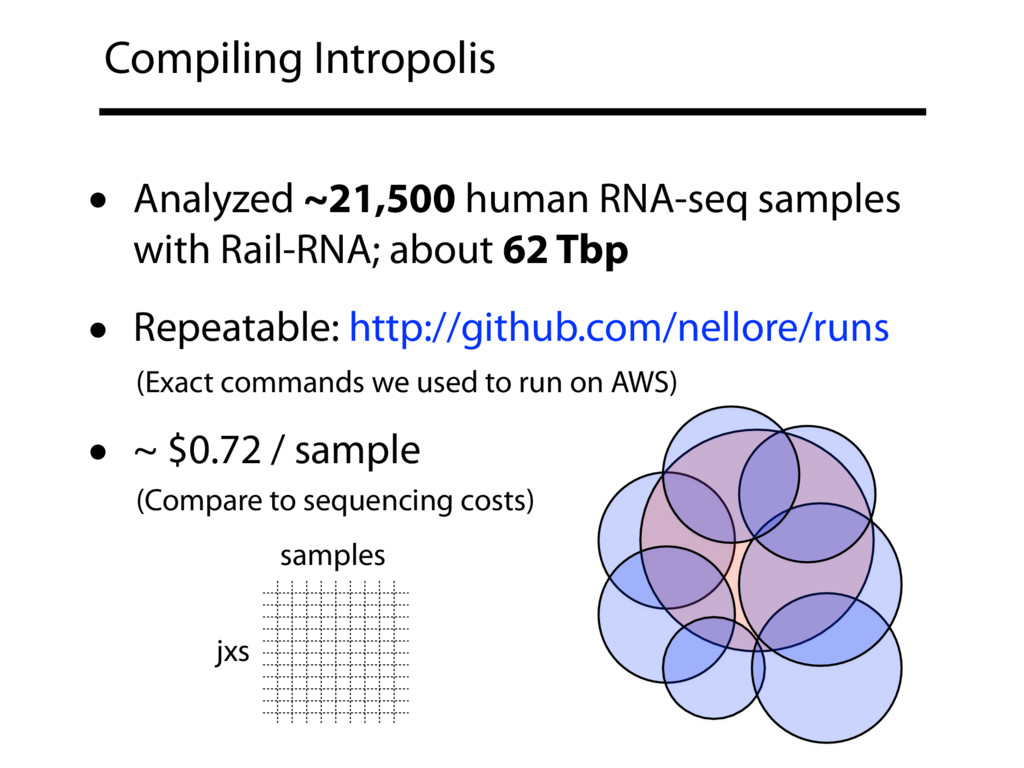{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}