recommendations Example project Bacterial epigenome characterization Kinetics in SMRT® Sequencing Analysis recommendations Where to find additional information
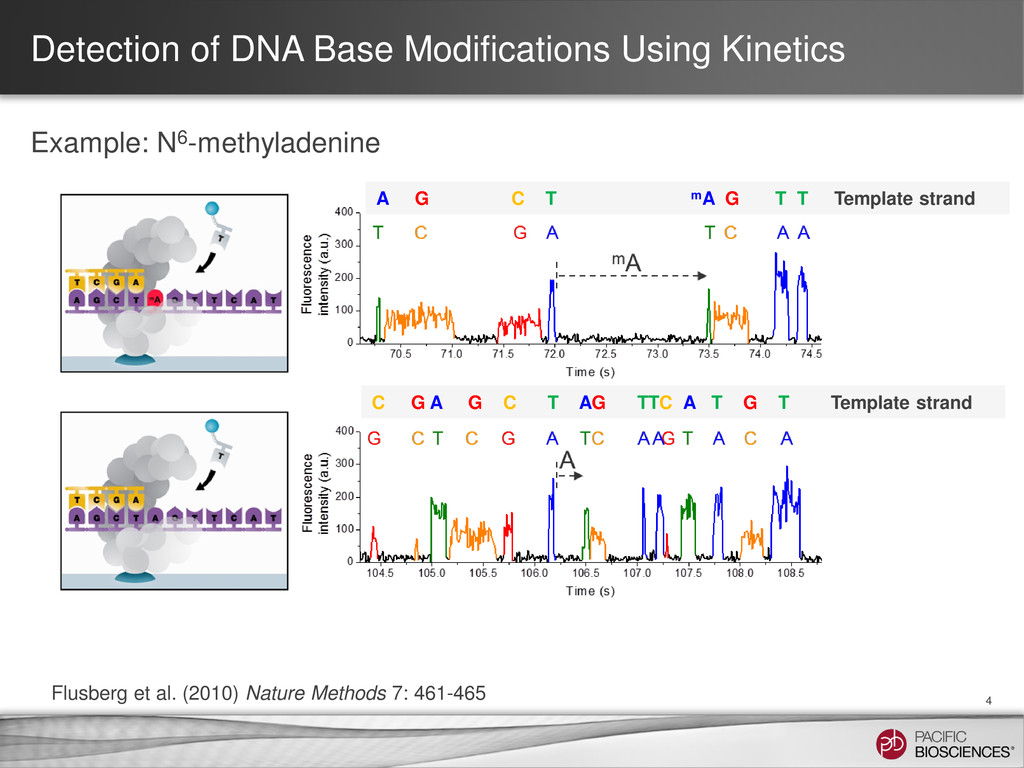
for each nucleotide addition to call bases • Same information can also be used to distinguish modified and native bases • Can compare results of SMRT Sequencing to an in silico kinetic reference for dynamics without modifications to infer the presence of bases different from A, C, G or T 3 Interpulse duration (IPD)
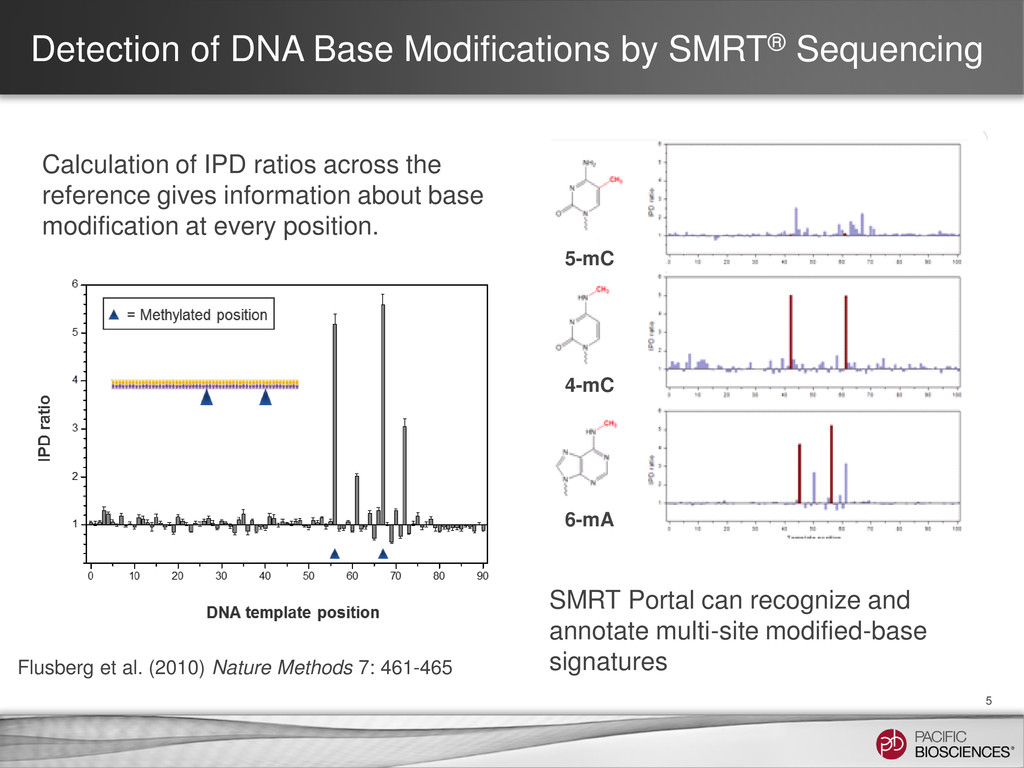
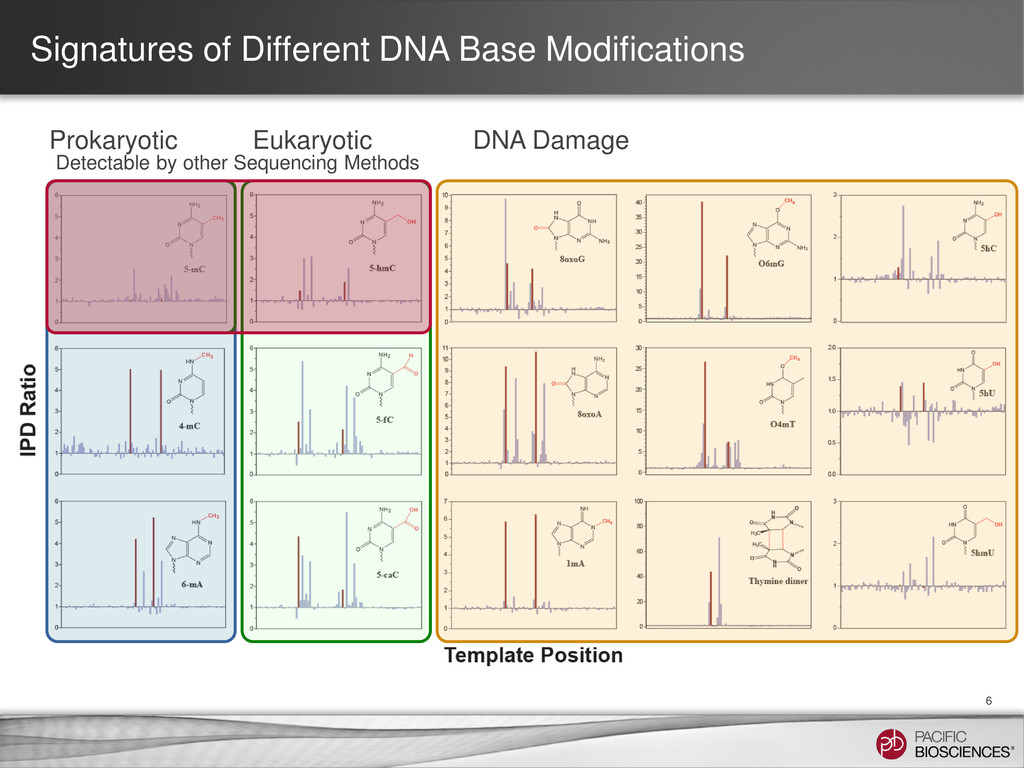
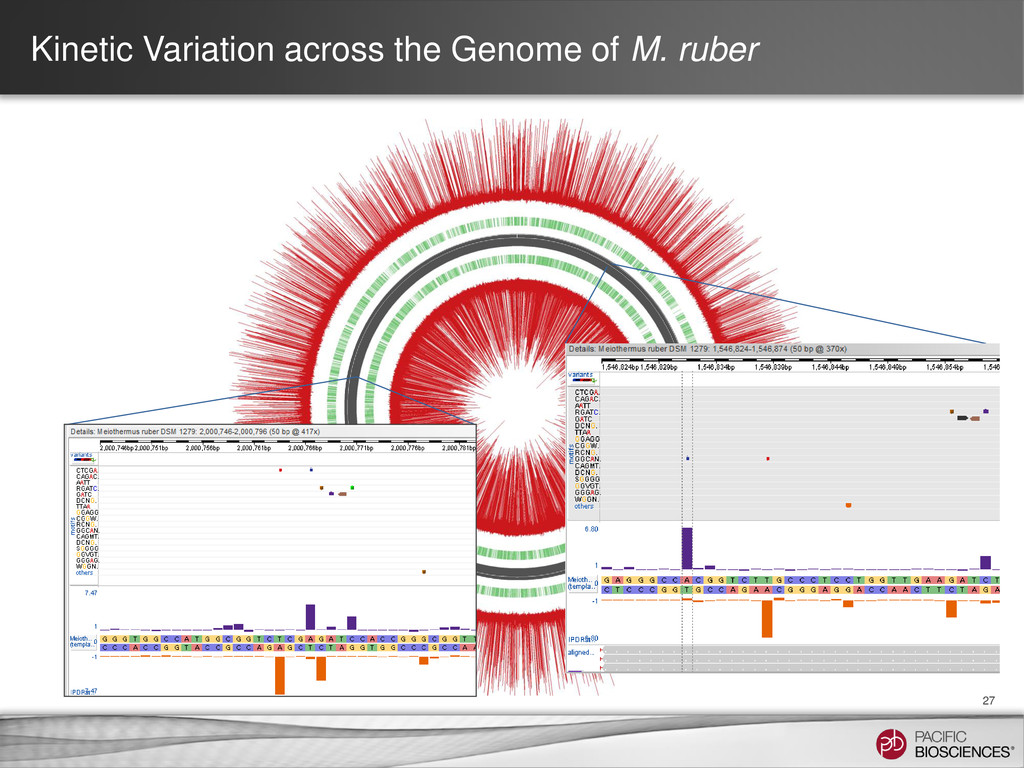
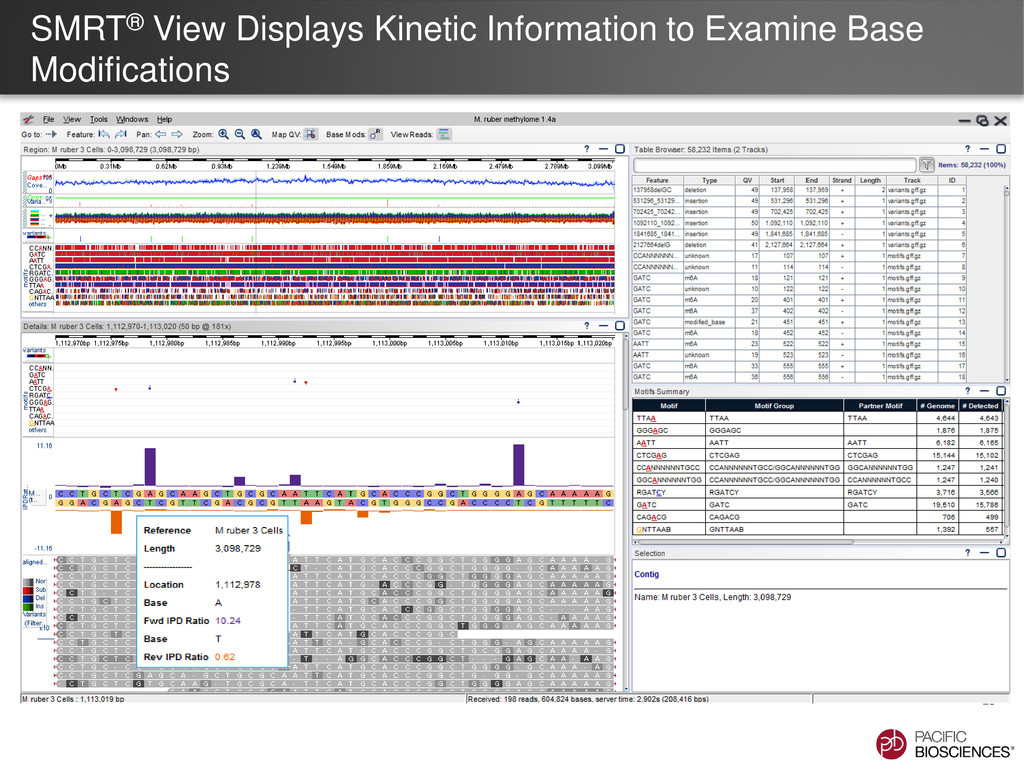
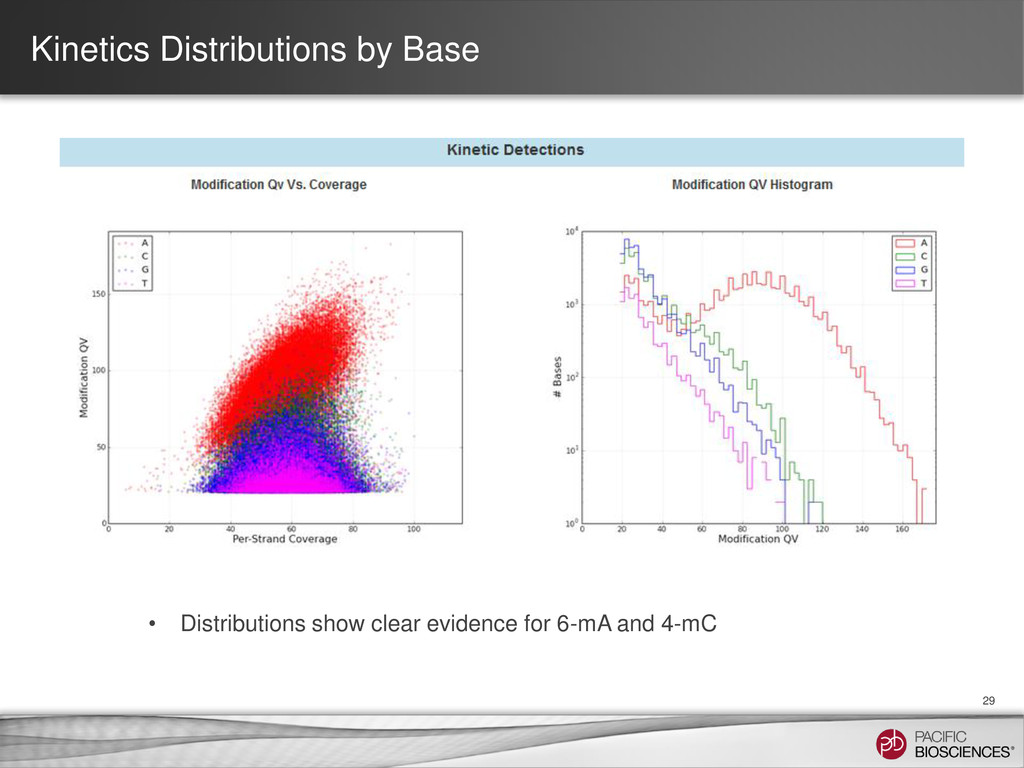
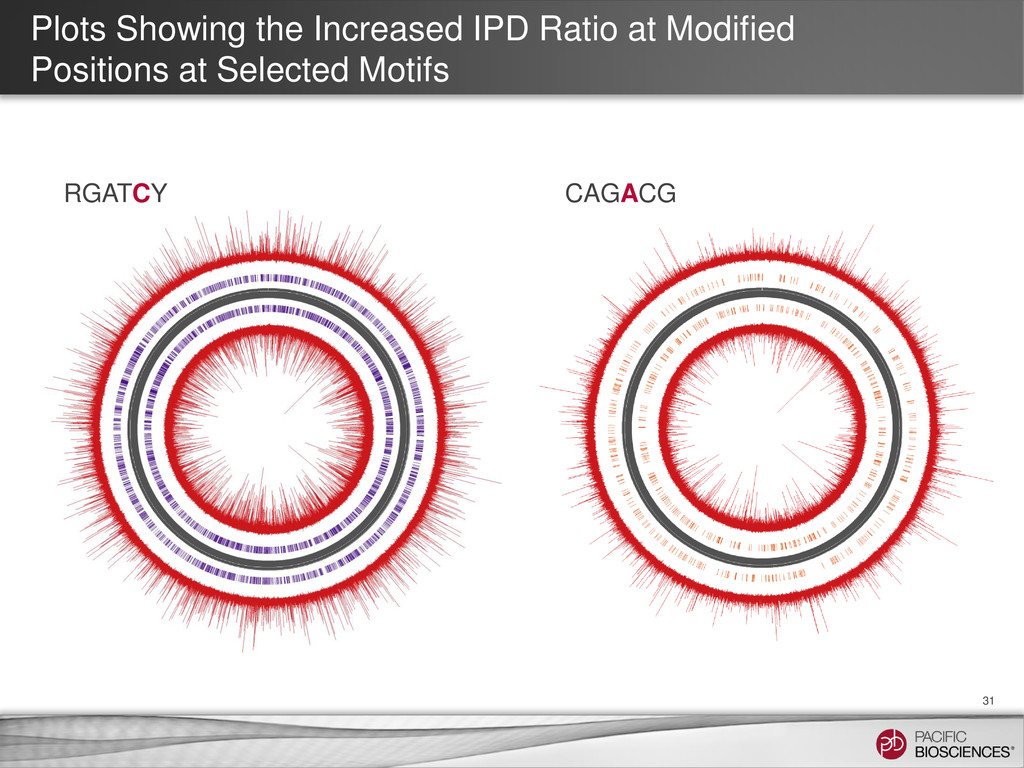
et al. (2010) Nature Methods 7: 461-465 SMRT Portal can recognize and annotate multi-site modified-base signatures 5-mC 4-mC 6-mA Calculation of IPD ratios across the reference gives information about base modification at every position.
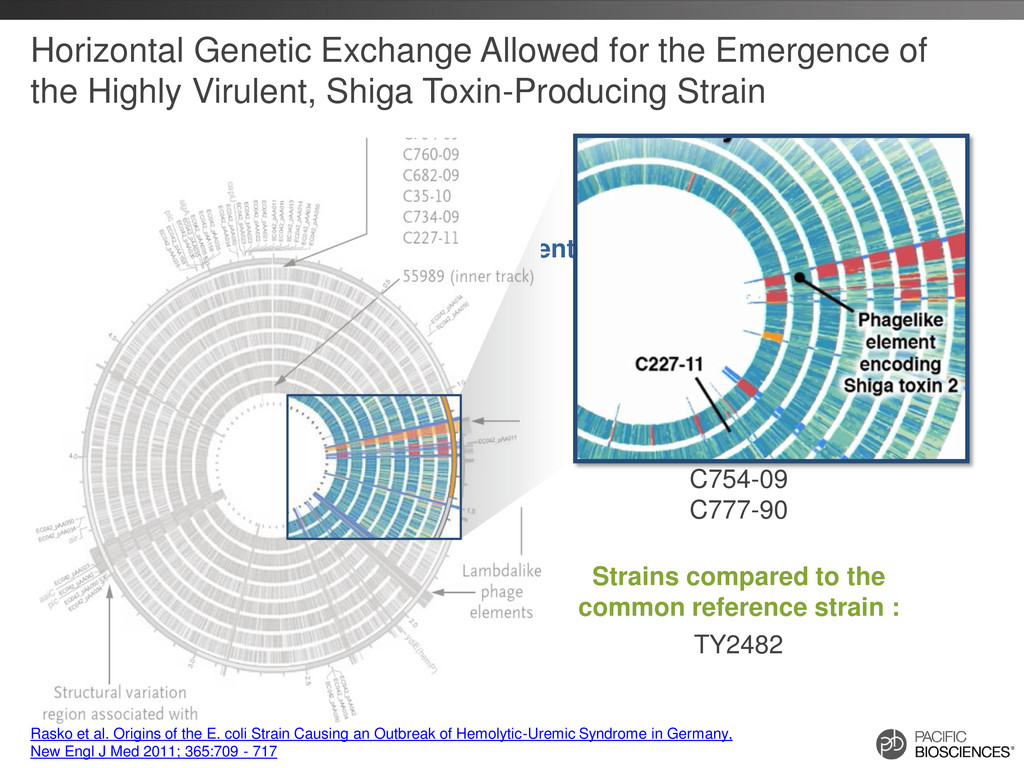
C35-10 C682-09 C760-09 C754-09 C777-90 Horizontal Genetic Exchange Allowed for the Emergence of the Highly Virulent, Shiga Toxin-Producing Strain Strains compared to the common reference strain : TY2482 Rasko et al. Origins of the E. coli Strain Causing an Outbreak of Hemolytic-Uremic Syndrome in Germany, New Engl J Med 2011; 365:709 - 717
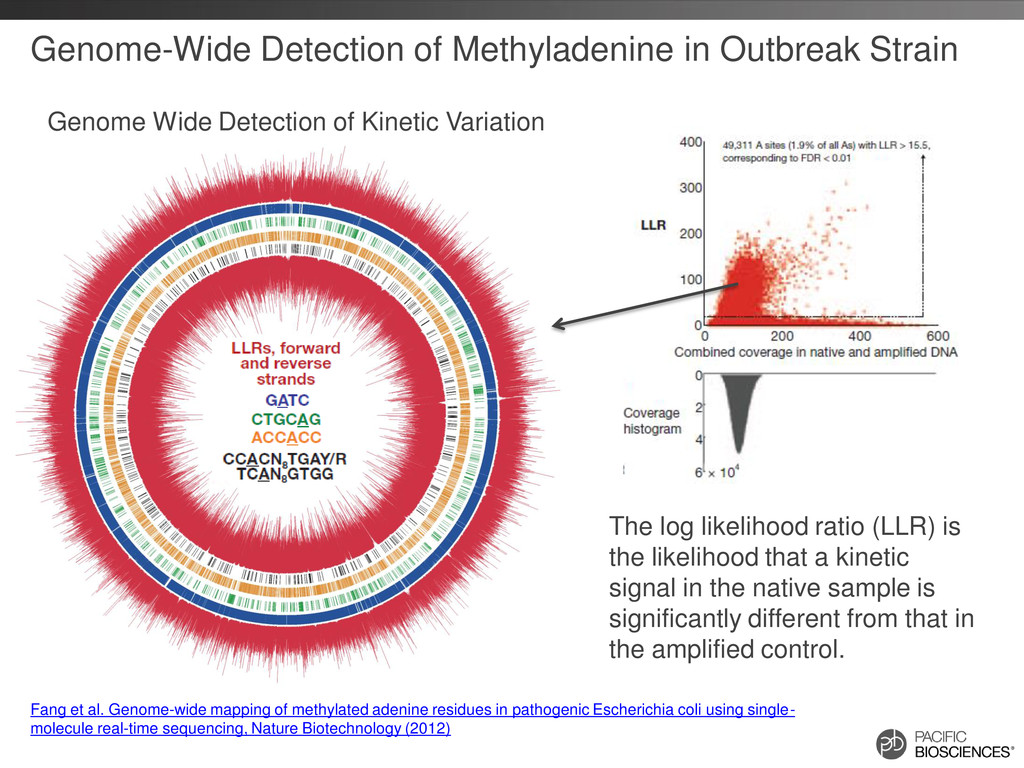
of Kinetic Variation The log likelihood ratio (LLR) is the likelihood that a kinetic signal in the native sample is significantly different from that in the amplified control. Fang et al. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single- molecule real-time sequencing, Nature Biotechnology (2012)
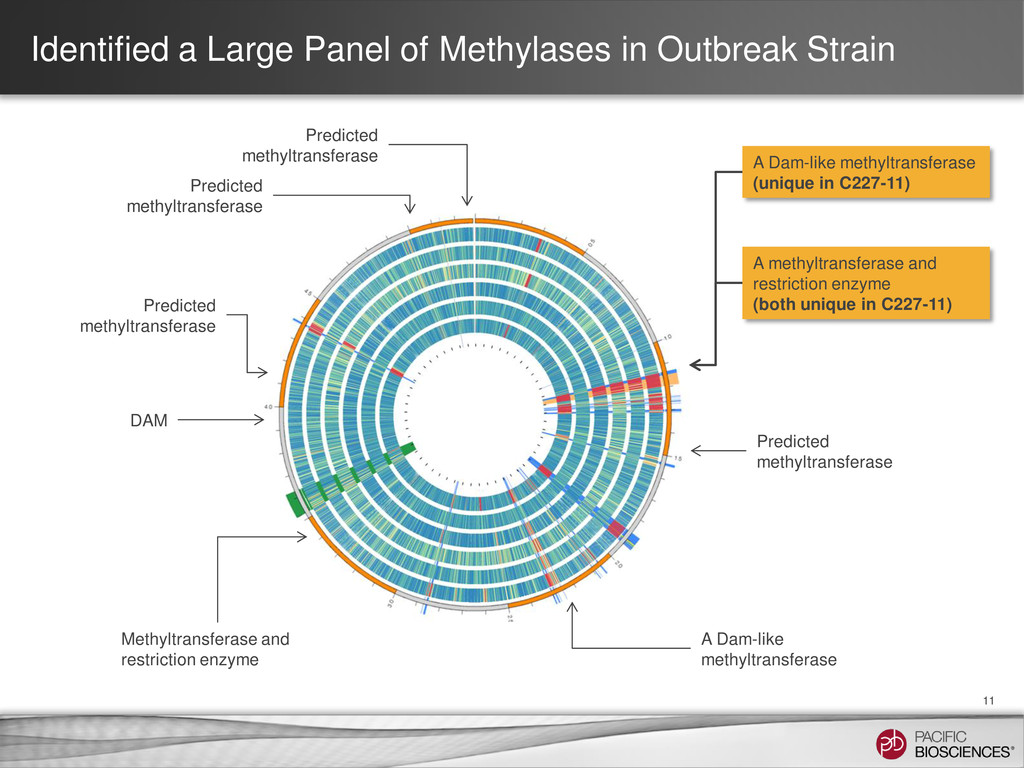
Predicted methyltransferase Predicted methyltransferase A methyltransferase and restriction enzyme (both unique in C227-11) A Dam-like methyltransferase (unique in C227-11) Predicted methyltransferase A Dam-like methyltransferase DAM Methyltransferase and restriction enzyme Predicted methyltransferase
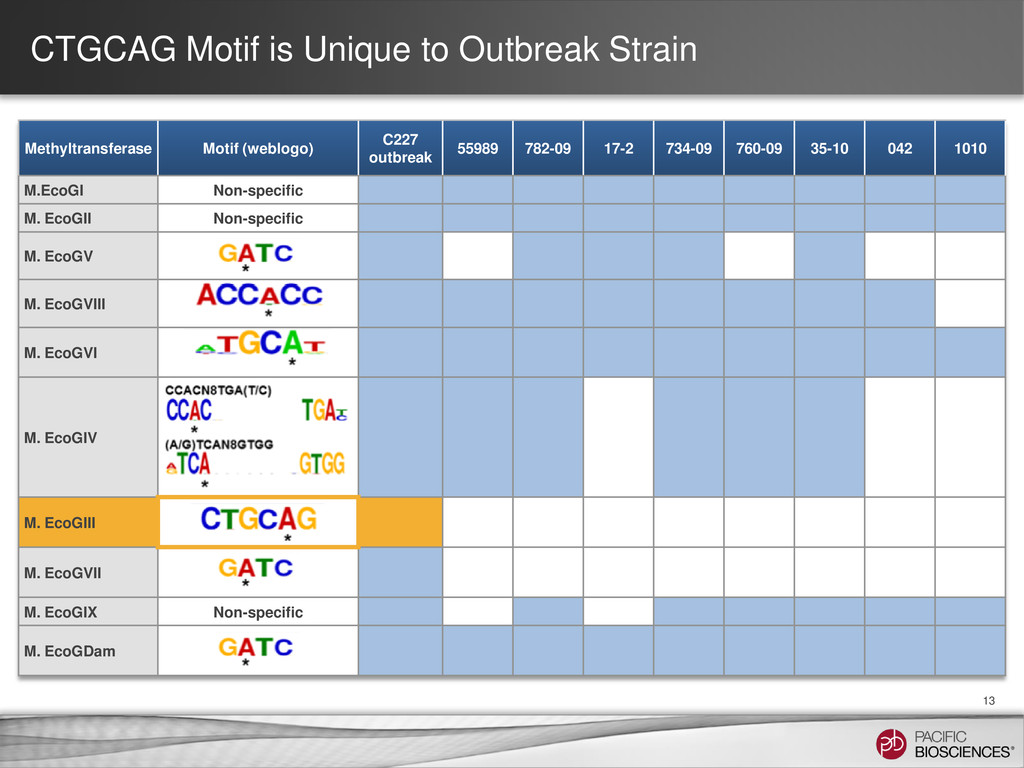
(weblogo) C227 outbreak 55989 782-09 17-2 734-09 760-09 35-10 042 1010 M.EcoGI Non-specific M. EcoGII Non-specific M. EcoGV M. EcoGVIII M. EcoGVI M. EcoGIV M. EcoGIII M. EcoGVII M. EcoGIX Non-specific M. EcoGDam
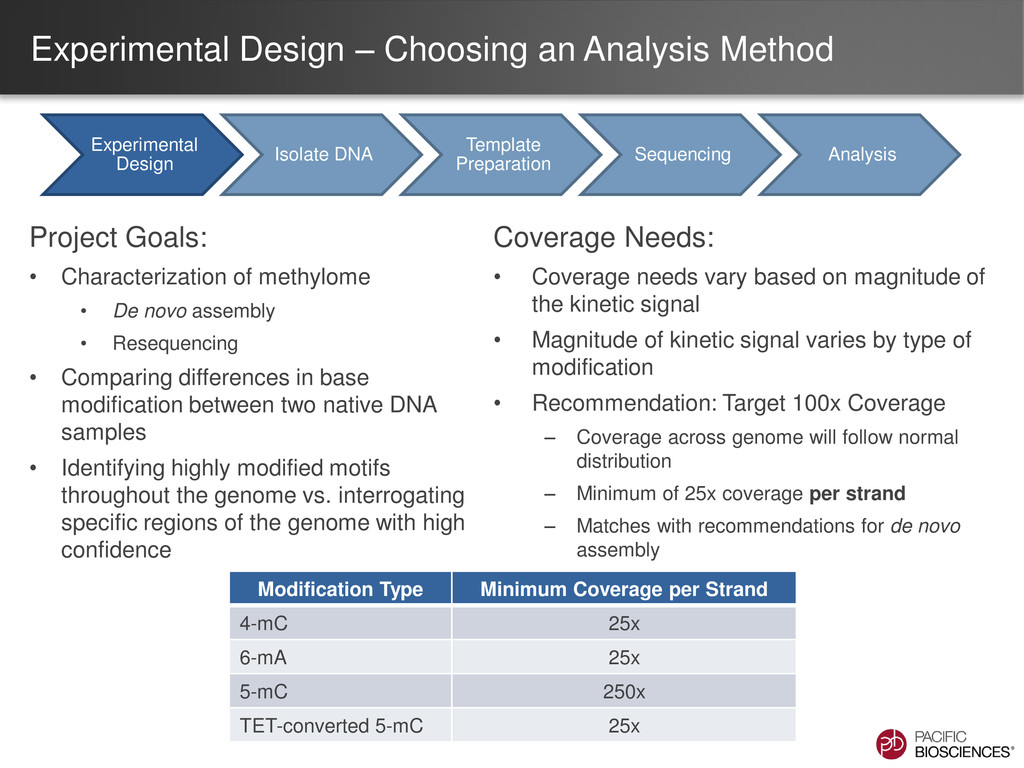
Characterization of methylome • De novo assembly • Resequencing • Comparing differences in base modification between two native DNA samples • Identifying highly modified motifs throughout the genome vs. interrogating specific regions of the genome with high confidence Coverage Needs: • Coverage needs vary based on magnitude of the kinetic signal • Magnitude of kinetic signal varies by type of modification • Recommendation: Target 100x Coverage – Coverage across genome will follow normal distribution – Minimum of 25x coverage per strand – Matches with recommendations for de novo assembly Experimental Design Isolate DNA Template Preparation Sequencing Analysis Modification Type Minimum Coverage per Strand 4-mC 25x 6-mA 25x 5-mC 250x TET-converted 5-mC 25x
Influence Experiment Planning 19 Control Options Definition In silico Computational model for predicting the mean IPD per given sequence context at the position of inquiry Amplified Control created by separately sequencing an amplified version of the sample of interest Native Comparison of two native DNA samples to identify differences in modifications present Advantages Eliminates need for data from an amplified control, reducing sequence data required May produce lower background (statistical noise) than in-silico control when sequencing to very high coverage Focuses on regions of differential modification When to Use • Default analysis mode • All polymerases supported • C2 chemistry only • For identification • XL chemistry • Confirming results from in-silico control Comparative studies to find differential modification
amplification – must use native DNA! – Except if using amplified control • DNA-damage repair – Does not impact methylated bases (4-mC, 6-mA, 5-mC) – Avoid for other types of modification (e.g., 8-oxo-G, dU) • Follow large-insert-library template-preparation recommendations for de novo assembly – Compatible with size-selection methods • Optional Tet1-conversion protocol on SampleNet • For resequencing, can use shorter-insert libraries – Proper mapping critical; insert size must exceed the repeat length – Multi-molecule analysis sufficient to perform motif analysis (exception rare DNA- damage events) Experimental Design Isolate DNA Template Preparation Sequencing Analysis
PacBio® RS II PacBio® RS II DNA Polymerase/ Binding Kit DNA/Polymerase Binding Kit P4 DNA/Polymerase Binding Kit P4 DNA Sequencing Kit DNA Sequencing Kit 2.0 (C2) DNA Sequencing Kit 2.0 (C2) Loading MagBead loading; follow protocol for insert size MagBead loading Stage Start Stage Start = yes Stage Start = no Movie Time 1 x 120 minute 1 x 45 movie or longer Experimental Design Isolate DNA Template Preparation Sequencing Analysis
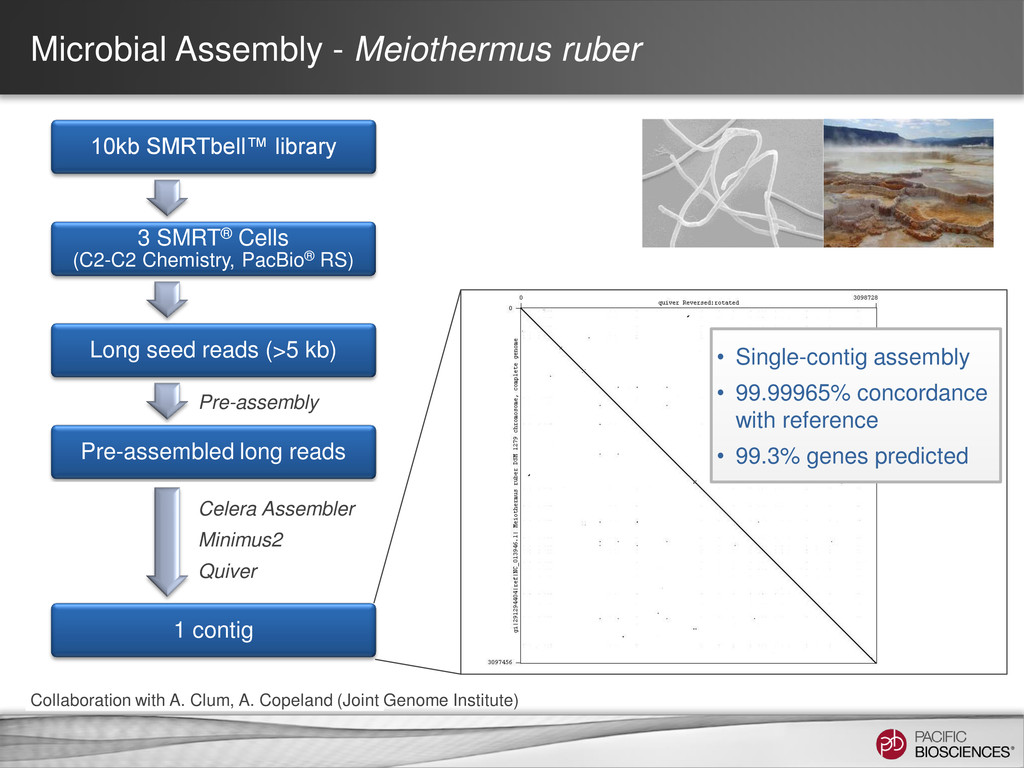
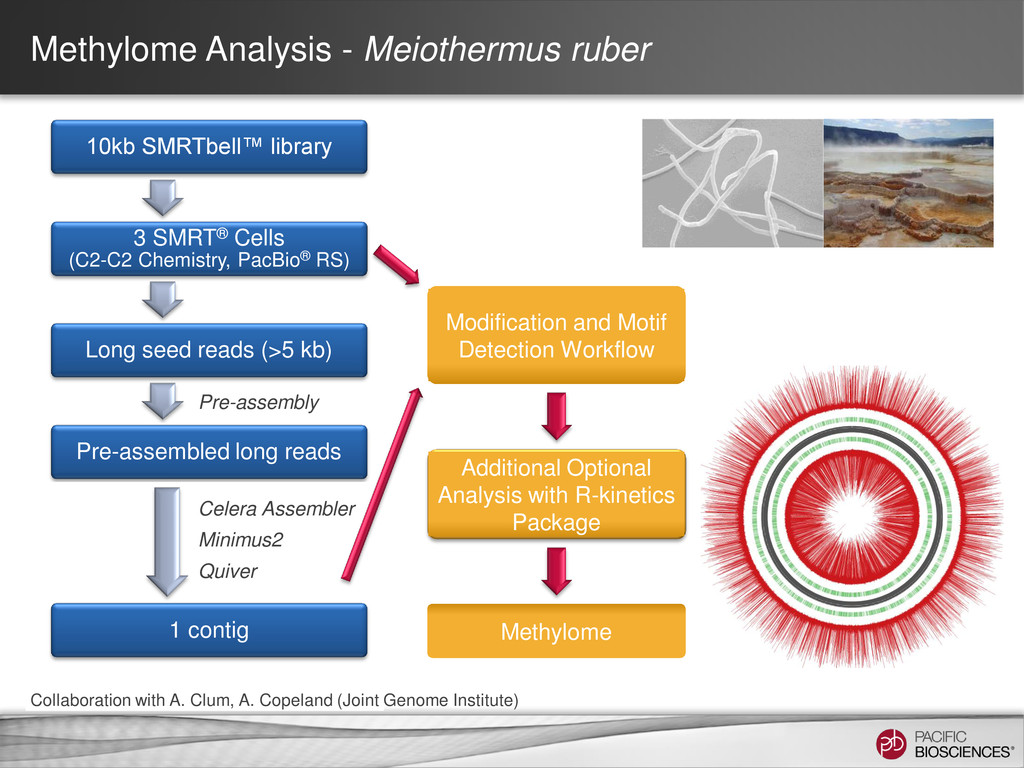
de novo Hierarchical Assembly • Upload Reference into SMRT® Portal • If using an amplified control • Align the WGA sequence to the reference sequence so that may be used as an amplified control • For P4-optimized in silico reference, use SMRT Portal v2.0.1 Identification of putative modification sites • SMRT Portal v2.0.1 using RS_Modification_and_Motif_Analysis.1 • If using an amplified control, refer to the amplified control job • View results in SMRT View Generate Reference Identification of putative sites with SMRT Portal v 2.0.1 Automated Motif Identification Visualize with SMRT View Experimental Design Isolate DNA Template Preparation Sequencing Analysis
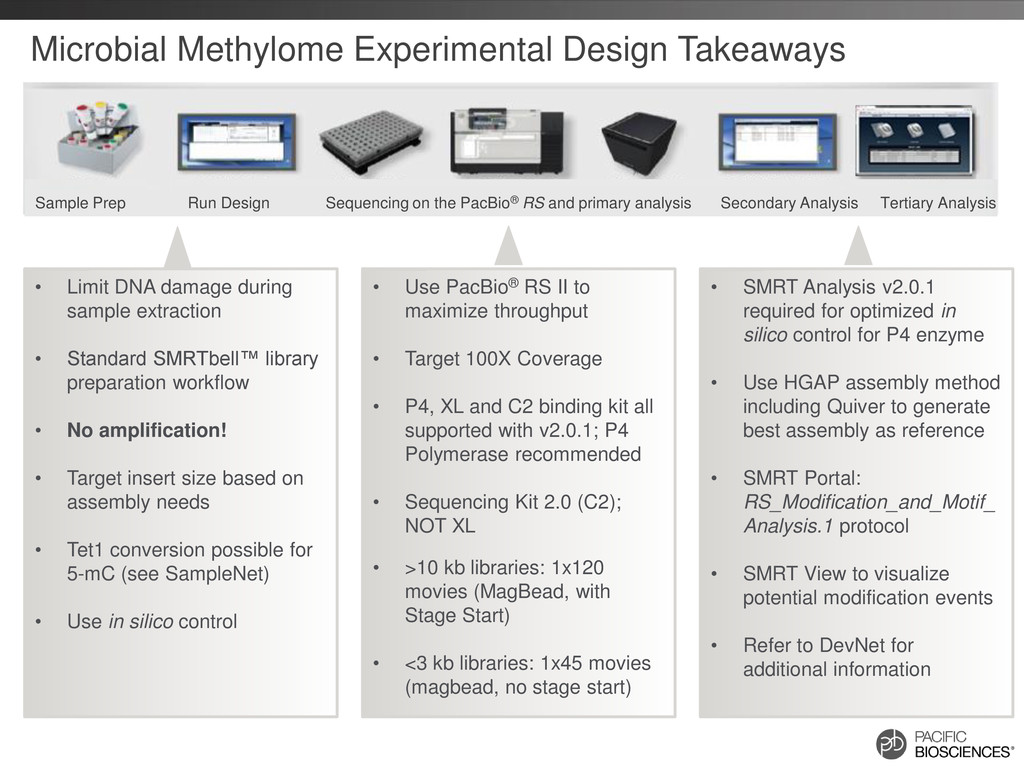
to maximize throughput • Target 100X Coverage • P4, XL and C2 binding kit all supported with v2.0.1; P4 Polymerase recommended • Sequencing Kit 2.0 (C2); NOT XL • >10 kb libraries: 1x120 movies (MagBead, with Stage Start) • <3 kb libraries: 1x45 movies (magbead, no stage start) • SMRT Analysis v2.0.1 required for optimized in silico control for P4 enzyme • Use HGAP assembly method including Quiver to generate best assembly as reference • SMRT Portal: RS_Modification_and_Motif_ Analysis.1 protocol • SMRT View to visualize potential modification events • Refer to DevNet for additional information • Limit DNA damage during sample extraction • Standard SMRTbell™ library preparation workflow • No amplification! • Target insert size based on assembly needs • Tet1 conversion possible for 5-mC (see SampleNet) • Use in silico control Sample Prep Run Design Sequencing on the PacBio® RS and primary analysis Secondary Analysis Tertiary Analysis
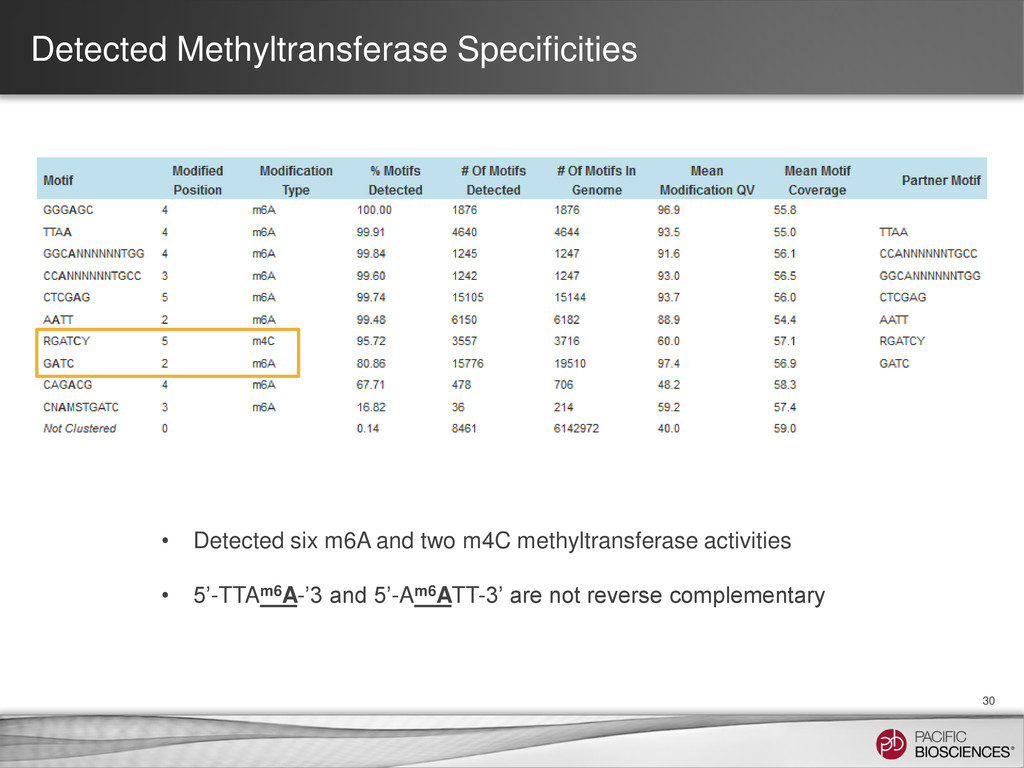
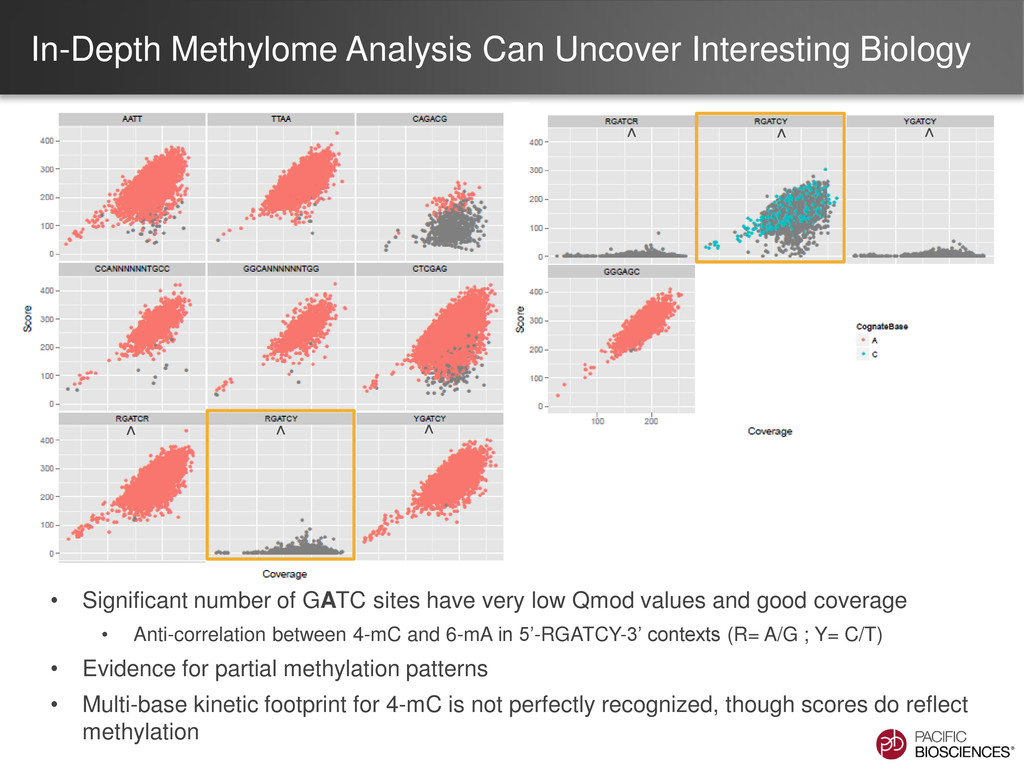
of GATC sites have very low Qmod values and good coverage • Anti-correlation between 4-mC and 6-mA in 5’-RGATCY-3’ contexts (R= A/G ; Y= C/T) • Evidence for partial methylation patterns • Multi-base kinetic footprint for 4-mC is not perfectly recognized, though scores do reflect methylation ^ ^ ^ ^ ^ ^
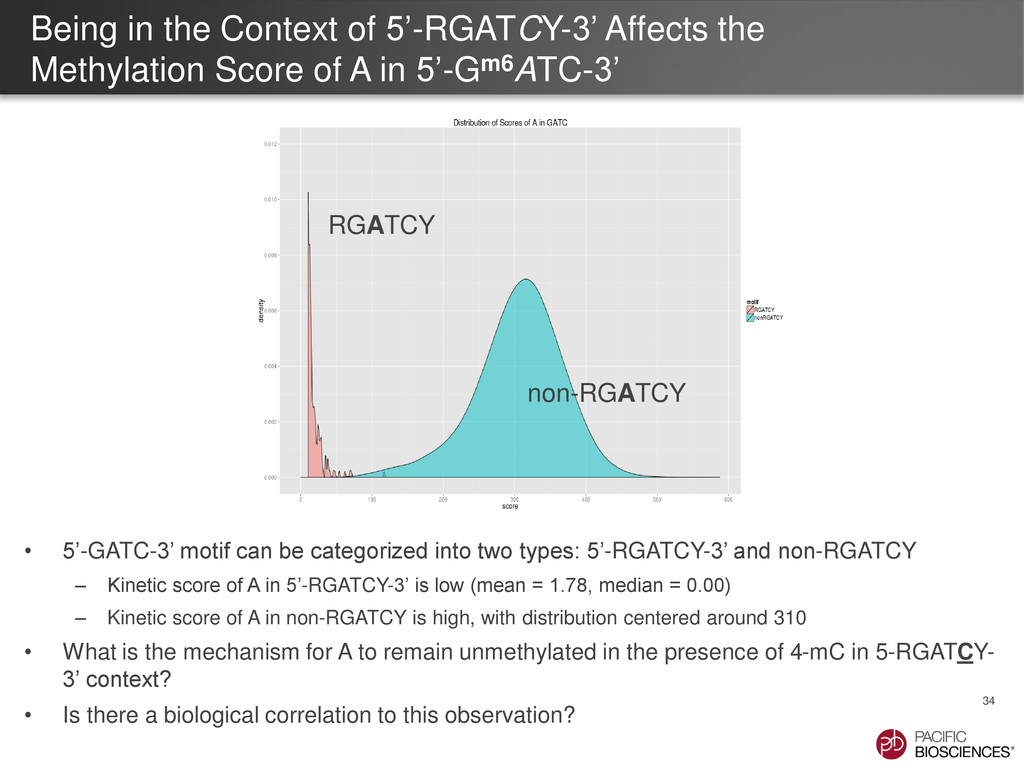
of A in 5’-Gm6ATC-3’ 34 • 5’-GATC-3’ motif can be categorized into two types: 5’-RGATCY-3’ and non-RGATCY – Kinetic score of A in 5’-RGATCY-3’ is low (mean = 1.78, median = 0.00) – Kinetic score of A in non-RGATCY is high, with distribution centered around 310 • What is the mechanism for A to remain unmethylated in the presence of 4-mC in 5-RGATCY- 3’ context? • Is there a biological correlation to this observation? non-RGATCY RGATCY
biochemical processes • SMRT® Sequencing provides a path to distinguishing dozens of different modifications • SMRT® Analysis simplifies detection of putative modification events using IPD-ratio analysis and a built-in motif-discovery algorithm • Guidance on experimental design and analysis recommendations available 36
paper – Overview of application area and relevant background • Webinars and Videos • Technical note – Bacterial-methylome analysis • Code and “How to” on DevNet – Tech note describing how to analyze IPDs – Motif analysis using R or GUI – Bacterial Assembly and Epigenetic Analysis Training Web Video http://www.pacificbiosciences.com/Tutorials/ Bacterial_Assembly_Epigenetic_Analysis_H GAP/story_html5.html • Publications • Base Modification Data – http://pacb.com/bmd http://pacb.com/basemod
are trademarks of Pacific Biosciences in the United States and/or other countries. All other trademarks are the sole property of their respective owners.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}