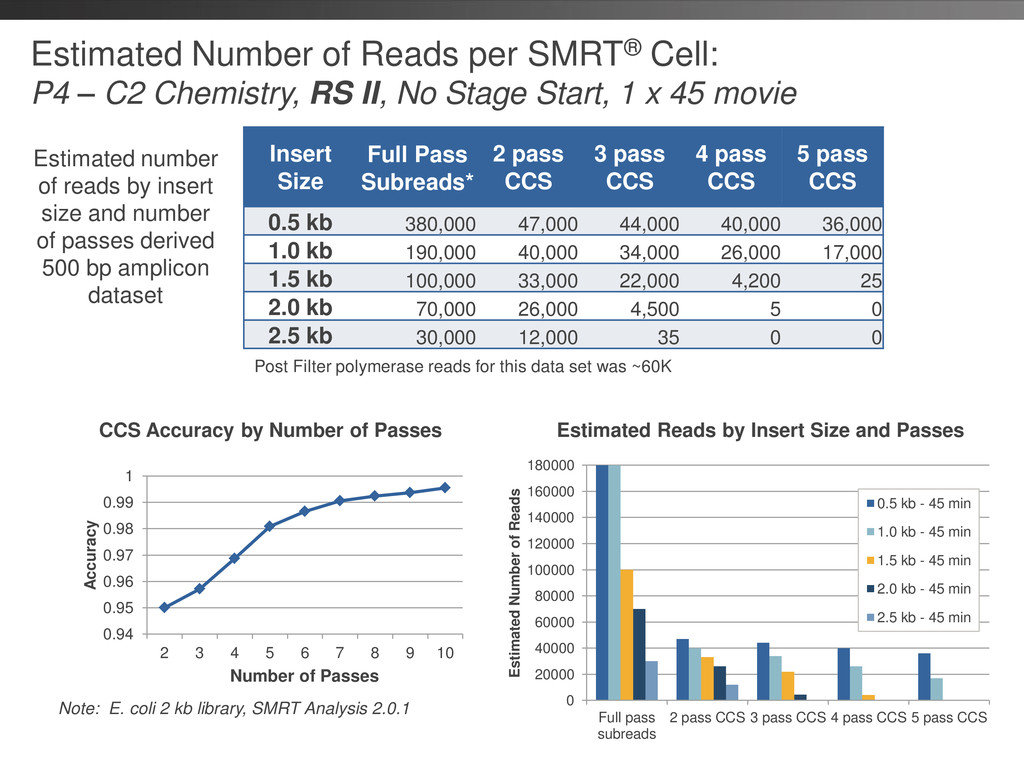
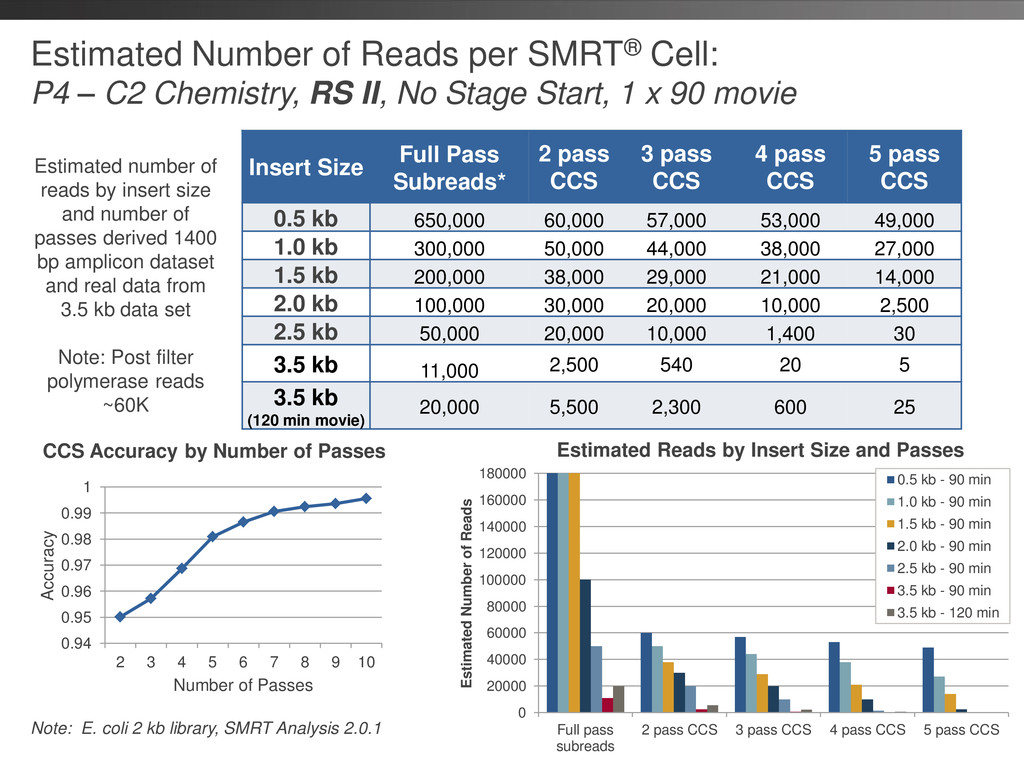
Chemistry, RS II, No Stage Start, 1 x 90 movie Insert Size Full Pass Subreads* 2 pass CCS 3 pass CCS 4 pass CCS 5 pass CCS 0.5 kb 650,000 60,000 57,000 53,000 49,000 1.0 kb 300,000 50,000 44,000 38,000 27,000 1.5 kb 200,000 38,000 29,000 21,000 14,000 2.0 kb 100,000 30,000 20,000 10,000 2,500 2.5 kb 50,000 20,000 10,000 1,400 30 3.5 kb 11,000 2,500 540 20 5 3.5 kb (120 min movie) 20,000 5,500 2,300 600 25 Estimated number of reads by insert size and number of passes derived 1400 bp amplicon dataset and real data from 3.5 kb data set Note: Post filter polymerase reads ~60K Note: E. coli 2 kb library, SMRT Analysis 2.0.1 0.94 0.95 0.96 0.97 0.98 0.99 1 2 3 4 5 6 7 8 9 10 Accuracy Number of Passes CCS Accuracy by Number of Passes Estimated Reads by Insert Size and Passes 0 20000 40000 60000 80000 100000 120000 140000 160000 180000 Full pass subreads 2 pass CCS 3 pass CCS 4 pass CCS 5 pass CCS Estimated Number of Reads 0.5 kb - 90 min 1.0 kb - 90 min 1.5 kb - 90 min 2.0 kb - 90 min 2.5 kb - 90 min 3.5 kb - 90 min 3.5 kb - 120 min
{kind=link}
{kind=link}
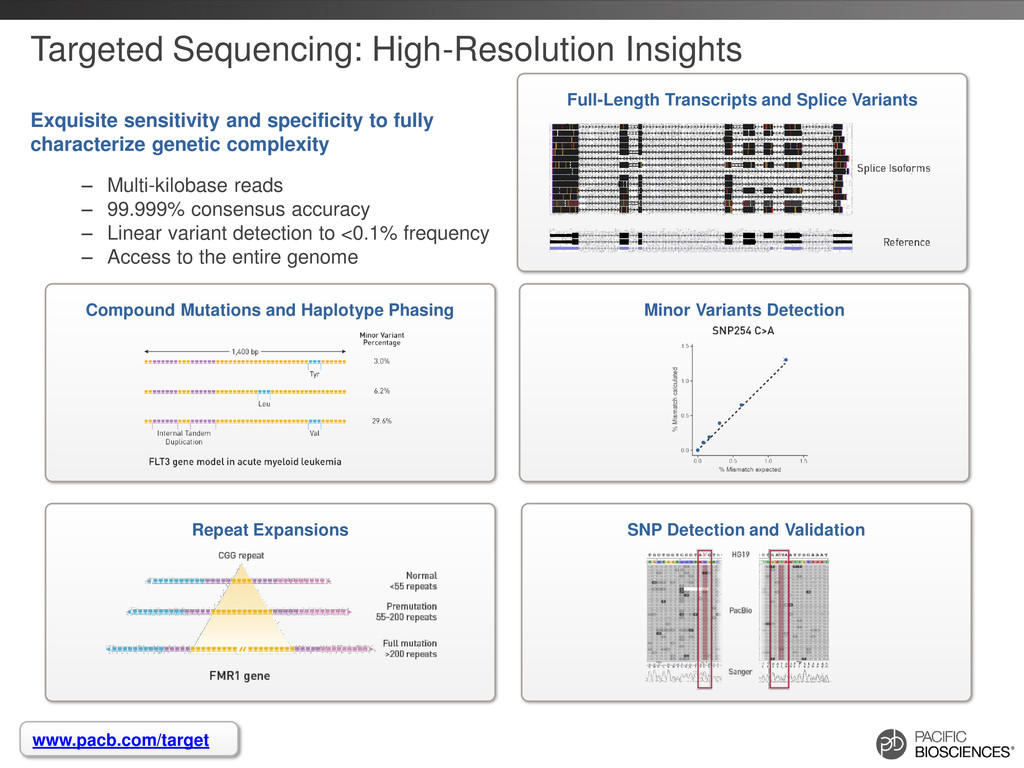{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
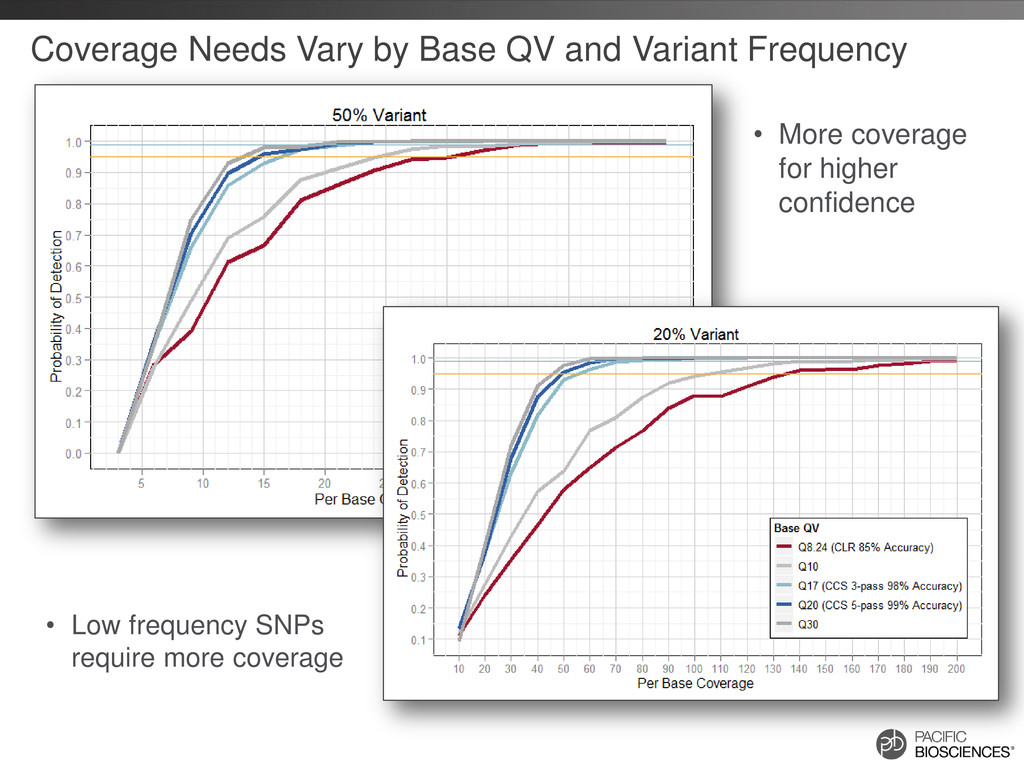{kind=link}
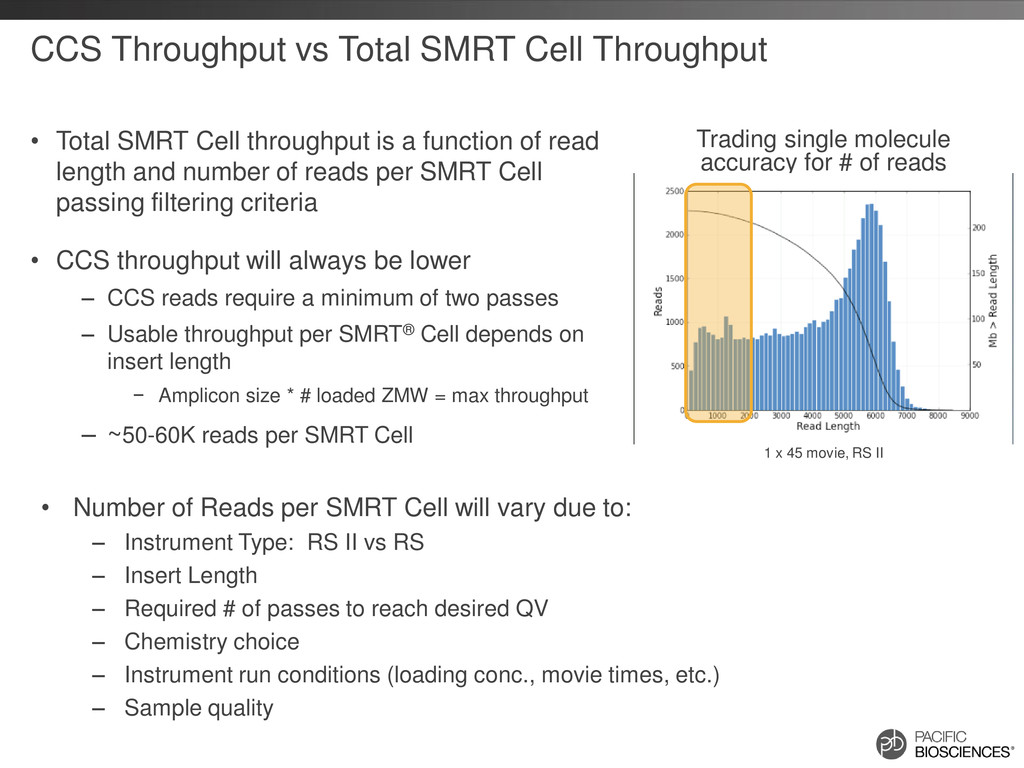{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
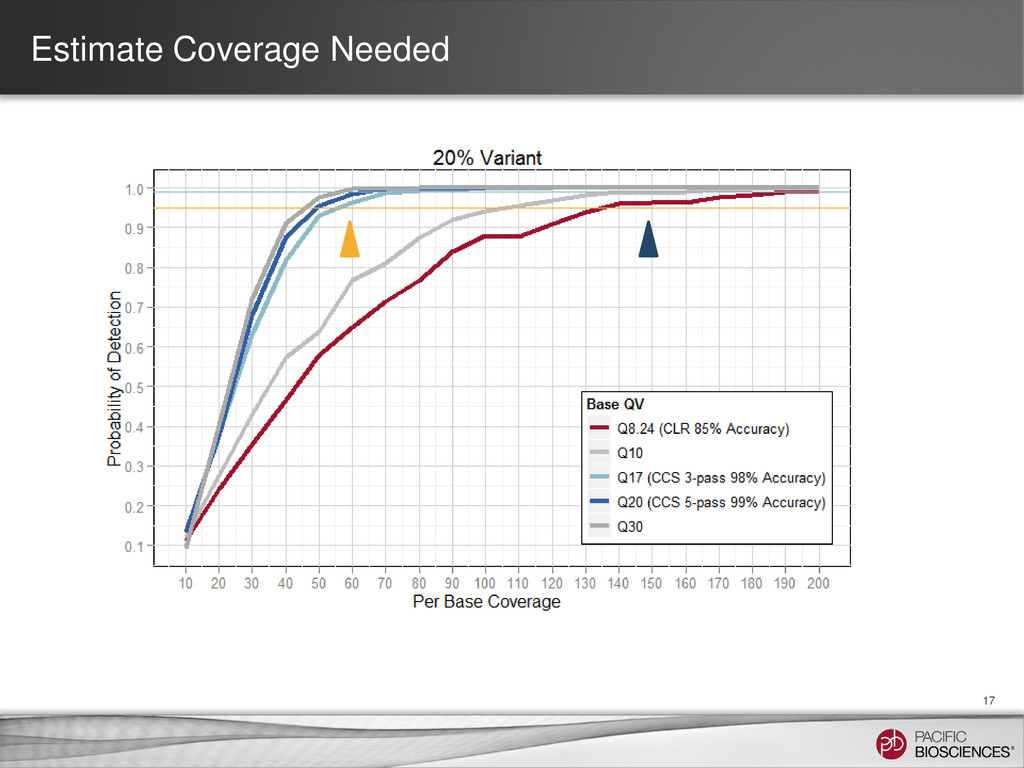{kind=link}
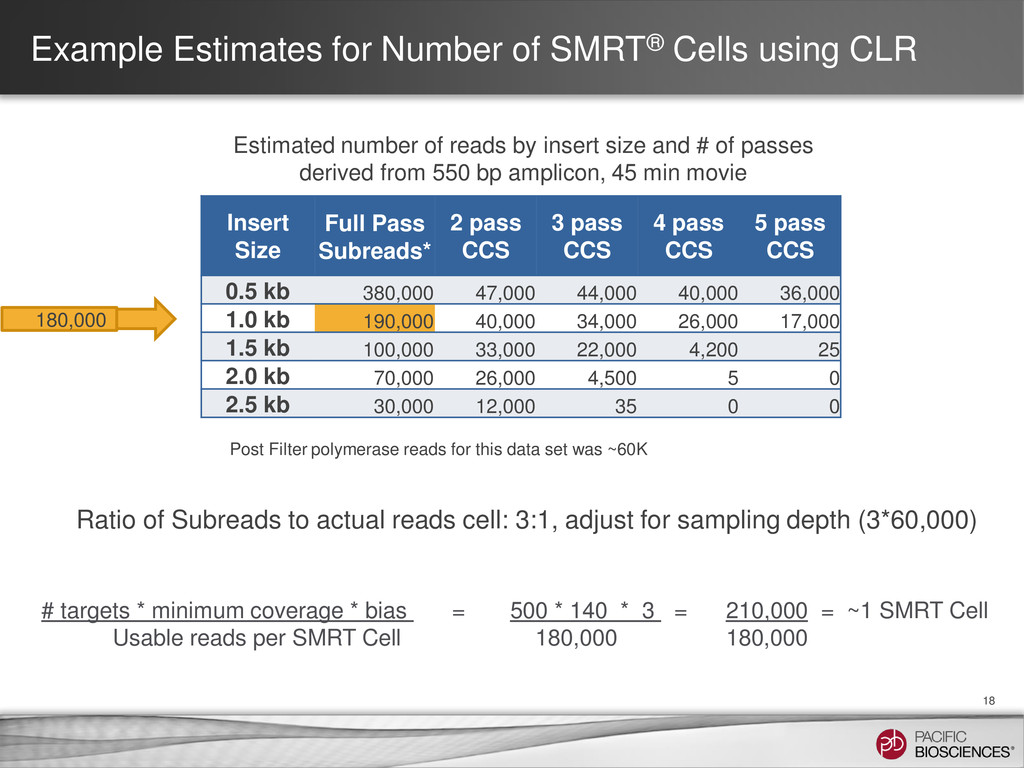{kind=link}
{kind=link}
{kind=link}
{kind=link}
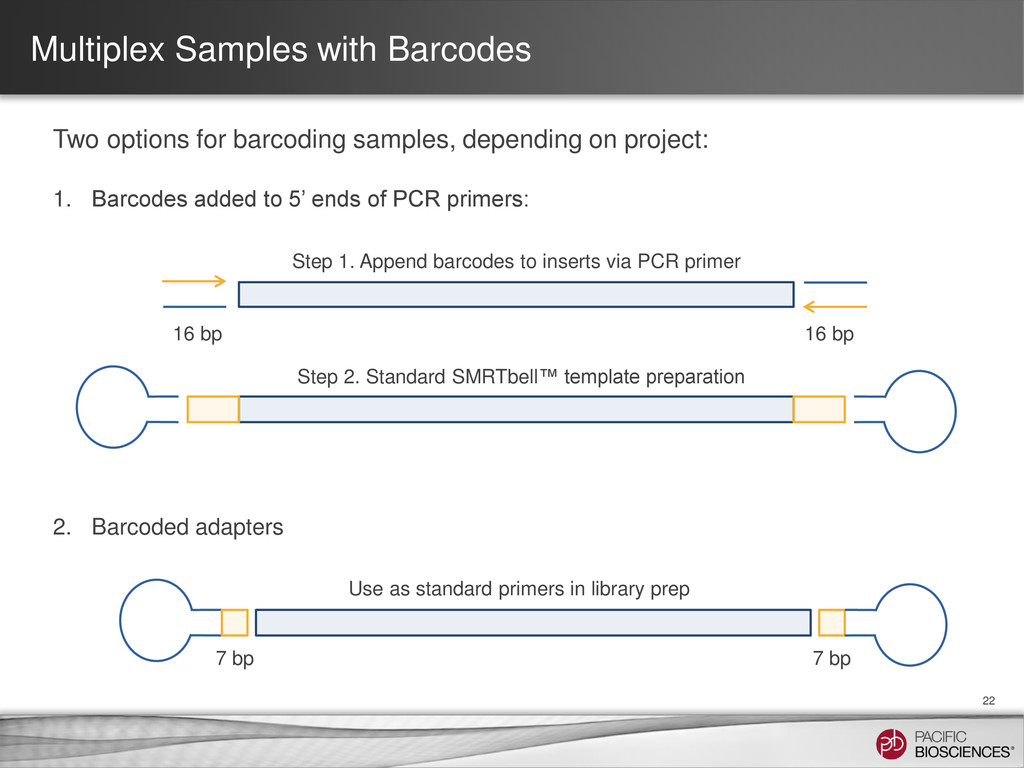{kind=link}
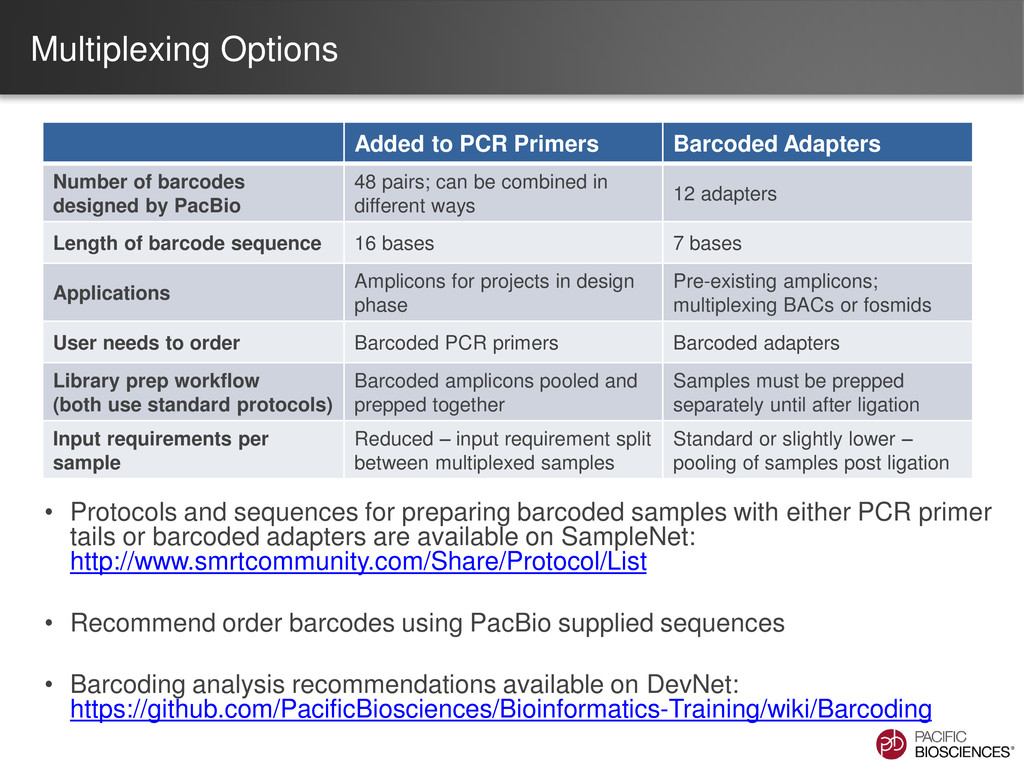{kind=link}
{kind=link}
{kind=link}
{kind=link}
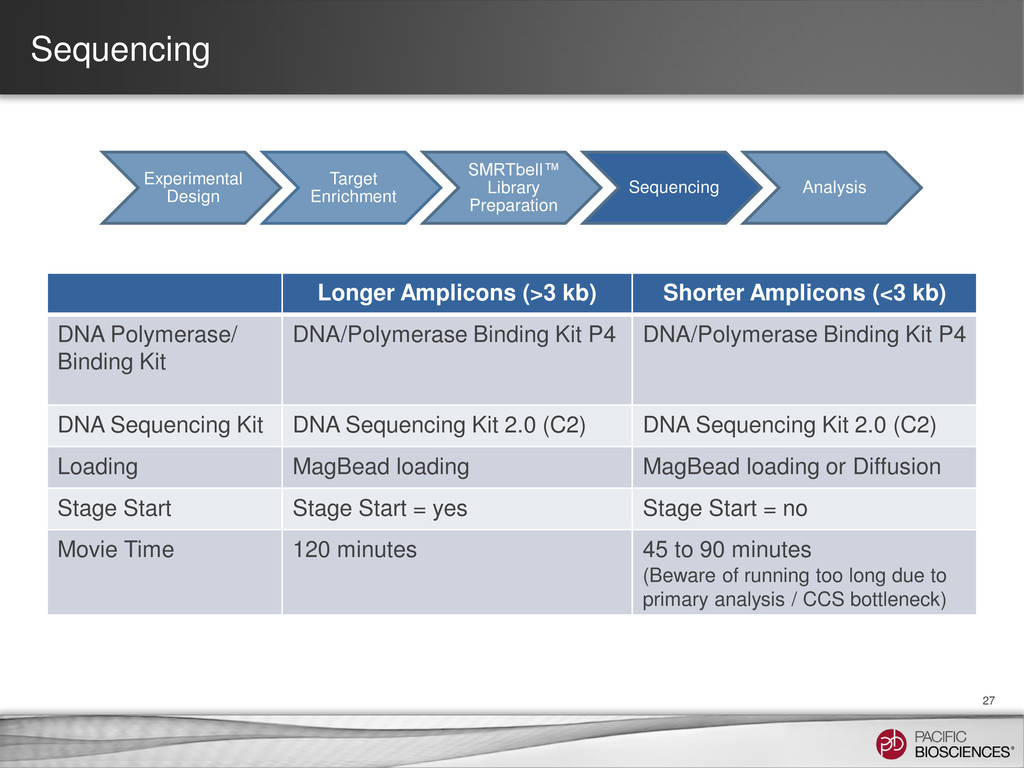{kind=link}
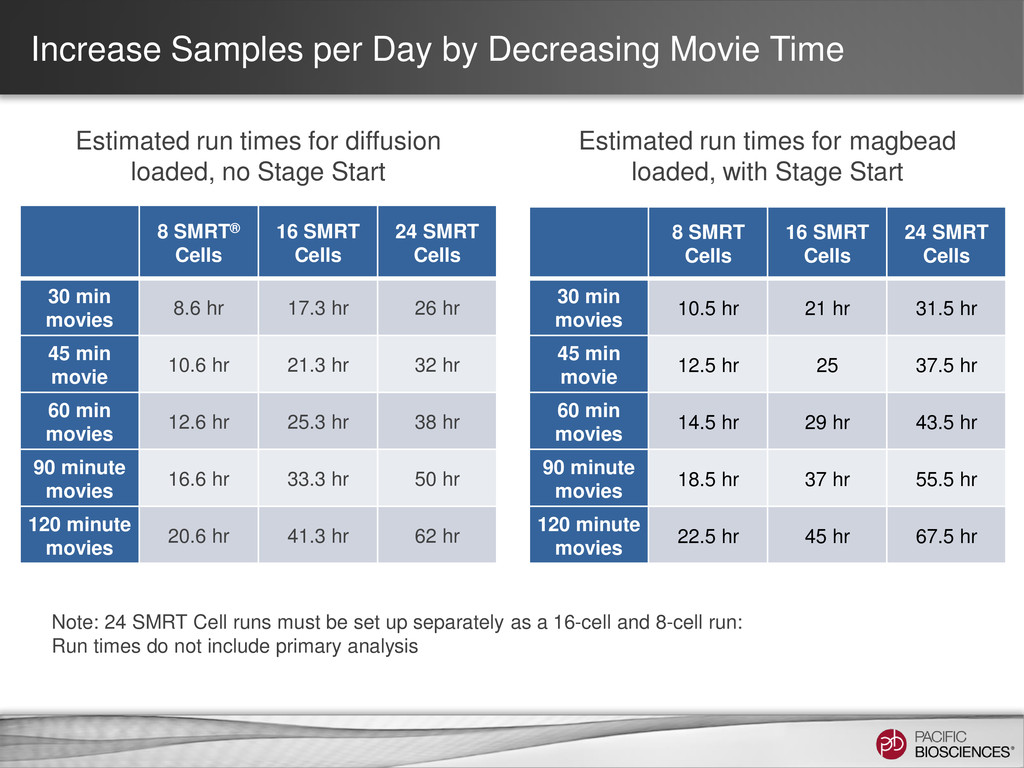{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}