body's connective tissue. Connective tissue holds together the body's cells, organs and tissues, as well as being important for growth and development. Connective tissue is made up of proteins. The protein that plays a role in Marfan syndrome is called fibrillin-1. Marfan syndrome is caused by a defect (or mutation) in the gene that tells the body how to produce fibrillin-1. This mutation results in an increase in a protiein called transforming growth fator beta (TGF-ß). The increase in TGF-ß causes problems in connective tissue throughout the body, These in turn create the features and medical issues associated with Marfan syndrome.
syndrome can in turn affect many aspects and systems of the body. Features are seen in the heart, blood vessels, bones, joints, eyes, lungs, skin, nervous system and eyes. Some of these features can be life-threatening. Marfan syndrome has no impact on intelligence. About 1 in 5,000 people have Marfan syndrome, including men and women of all ethnicities (0.02% of the population). For comparitive prevalence of some other genetic disorders; Hypermobile Ehlers-Danlos syndrome (hEDS); 1 in 5,000, cystic fibrosis; 1 in 1250, Huntingtons disease; 1 in 15,000. 75% of Marfan syndrome is inherited, with 25% a spontaneous mutation. The likelihood of an individual with Marfan syndrome passing this on to a child is 50%.
features may not become evident immediately. Some will have Marfan features at birth or as young children, but others will only become apparent as they develop issues in adulthood. Some of these features become more problematic over time. It's estimated that almost 50% of individuals with Marfan syndrome don't know that they have it.
combination of features, some of which are easier to see. These include; Long arms, legs & fingers. Tall and thin body type. Curved spine. Chest that sinks in or sticks out. Flexible joints. Flat feet. Crowded teeth. Stretch marks unrealted to weight loss or gain.
include; Heart problems - especially those related to the aorta. Sudden pneumothorax. Eye problems such as severe nearsightedness, dislocated lens, detached retina, early glaucoma and early cataracts. Specific tests are required to detect these features.
Cardiovascular; HOW ARE THE BODY'S SYSTEMS AFFECTED? Long limbs. Tall & thin body type. Long digits. Curvature of the spine (scoliosis or kyphosis) Chest sinking in or sticking out (pectus excavatum or carinatum.) Flexible joints. Flat feet. High arched palate. Crowded teeth. Skeletal; Severe nearsightedness. Dislocated lens or retina. Early glaucoma or cataracts. Ocular; Spontaneous pneumothorax. Emphysaema. Asthma. Sleep apnoea. Respiratory; Stretch marks. Swelling of the sac around the spinal column (dural ectasia). Other; Abdomenal; Abdominal aortic aneurysm/rupture.
family member who may have suffered an early, unexplained, heart-related death. A complete physical examination. Echocardiogram. ECG. Ocular examination. CT or MRI of the lower back.
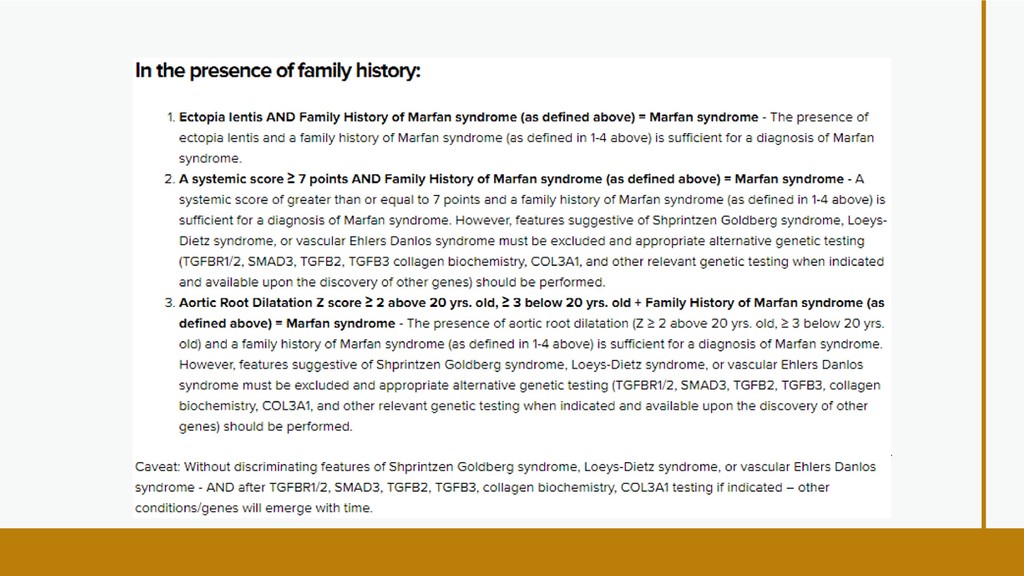
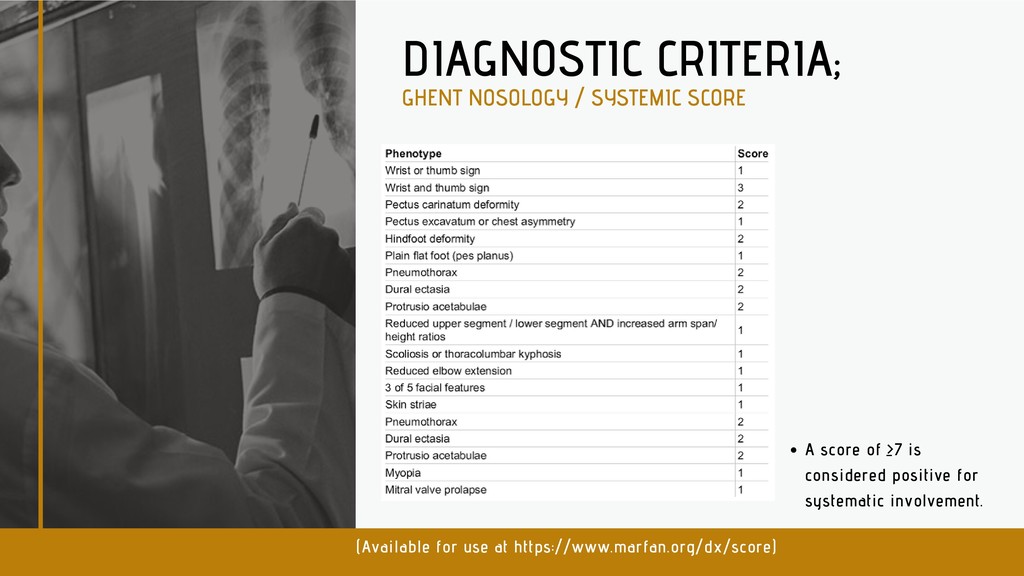
international panel of experts revised the criteria in 2010. Aortic root aneurysm and ectopia lentis (dislocated lenses) are now cardinal features. In the absence of any family history, the presence of these two features is sufficient for the unequivocal diagnosis of Marfan syndrome. In the absence of one of these two cardinal features, the presence of an FBN1 mutation or positive systemic score is required. In some cases, genetic testing can be helpful. Experts expect that, while use of new diagnostic criteria makes a definitive diagnosis of Marfan syndrome take longer, it decreases the risk of premature or missed diagnosis and facilitates a worldwide discussion of risk and management guidelines. DIAGNOSTIC CRITERIA;
it is possible to live a long, full life with proper treatment and management of symptoms. Every individual is different, depending on how severe or mild their features are. Various specialists will likely need to be involved in an individual's care - including a regular/annual echocardiogram to monitor the aorta size - perhaps with a more generalist doctor who will coordinate their input. Individuals may require a range of input from simple supportive therapies such as physiotherapy or counselling, to invasive surgery of the spine, eyes or aorta. Medications for chronic pain, blood- pressure control or anti-depressants may be helpful/necessary. High-inpact/intensity sports should be avoided. MANAGEMENT;
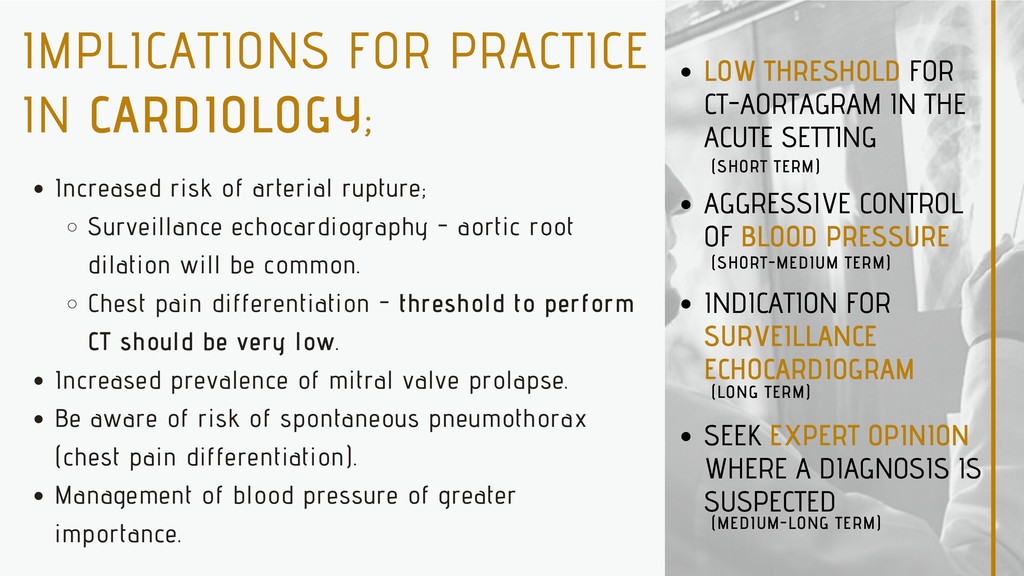
the dilation of the aorta and subsequent risk of aortic dissection (thoracic or abdominal). This is a life-threatening emergency and the most common cause of death in individuals with Marfan syndrome. In the emergency setting it's important that health professionals identify early on if there is a diagnosis (or suspicion or family history) of Marfan syndrome. Aortic dissection can be identified or excluded by CT scan. Some individuals with aortic dissection will require urgent surgery, whilst others will have intravenous drugs to aggressively control their blood pressure. Mortality is high (~20% prehospital, 1-3% per hour in the first 24hrs).
Surveillance echocardiography - aortic root dilation will be common. Chest pain differentiation - threshold to perform CT should be very low. Increased prevalence of mitral valve prolapse. Be aware of risk of spontaneous pneumothorax (chest pain differentiation). Management of blood pressure of greater importance. LOW THRESHOLD FOR CT-AORTAGRAM IN THE ACUTE SETTING AGGRESSIVE CONTROL OF BLOOD PRESSURE INDICATION FOR SURVEILLANCE ECHOCARDIOGRAM SEEK EXPERT OPINION WHERE A DIAGNOSIS IS SUSPECTED (MEDIUM-LONG TERM) (LONG TERM) (SHORT-MEDIUM TERM) (SHORT TERM)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}