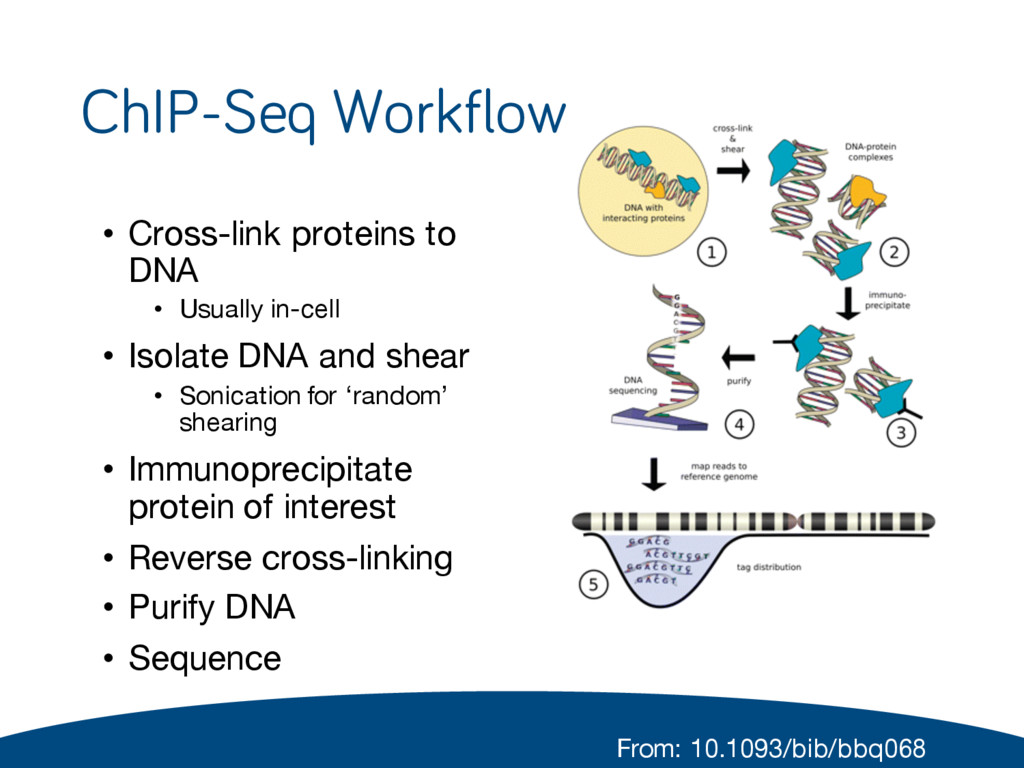
• Isolate DNA and shear • Sonication for ‘random’ shearing • Immunoprecipitate protein of interest • Reverse cross-linking • Purify DNA • Sequence From: 10.1093/bib/bbq068
Poisson process* • Deviations from Poisson define peak regions • Reads on either strand will flank the true binding position * Not really, due to multiple biases – but close enough… From: 10.1038/nmeth.1246
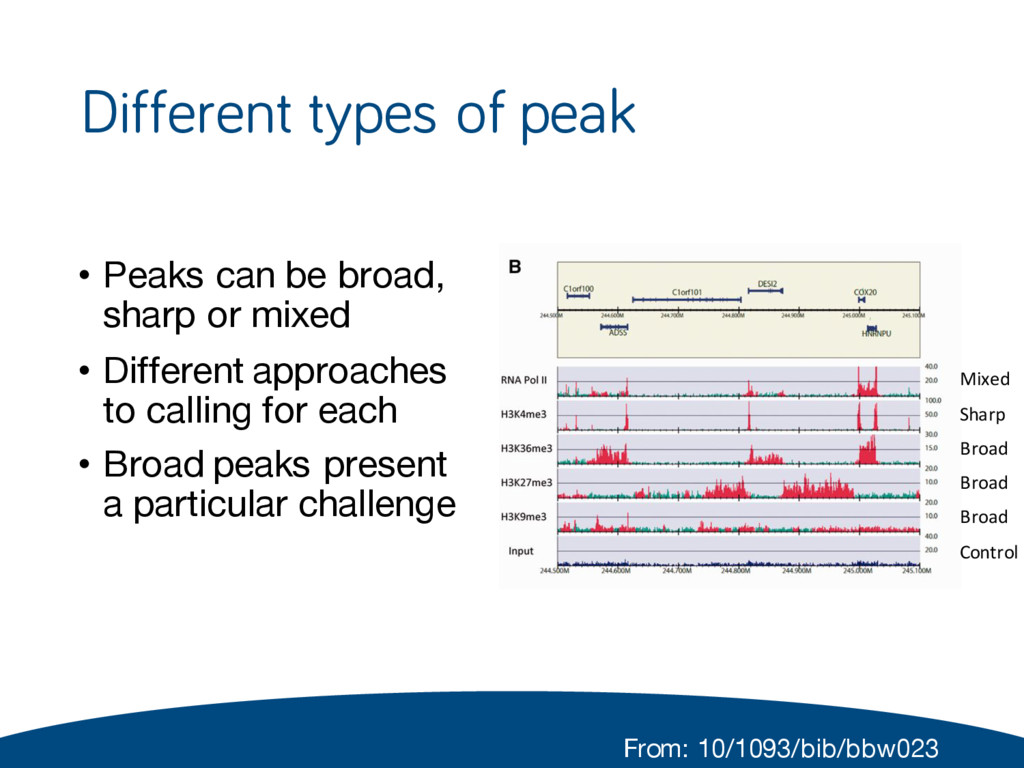
or mixed • Different approaches to calling for each • Broad peaks present a particular challenge Mixed Sharp Broad Broad Broad Control From: 10/1093/bib/bbw023
most not ‘fixable’ with bioinformatics • Major problems include: • Variable mappability of genome • Poor signal:noise ratio (S/N) from ChIP • Issues with low library complexity • Inappropriate or insufficient controls (and replicates)
obvious problems with regions of low mappability (repeats etc) • If these regions are of interest, a specific strategy needs to be taken • Paired reads, long reads, include multi-mapped in analysis? • ENCODE black-list regions of particular problems • Available for a few organisms • https://sites.google.com/site/anshulkundaje/projects/blacklists
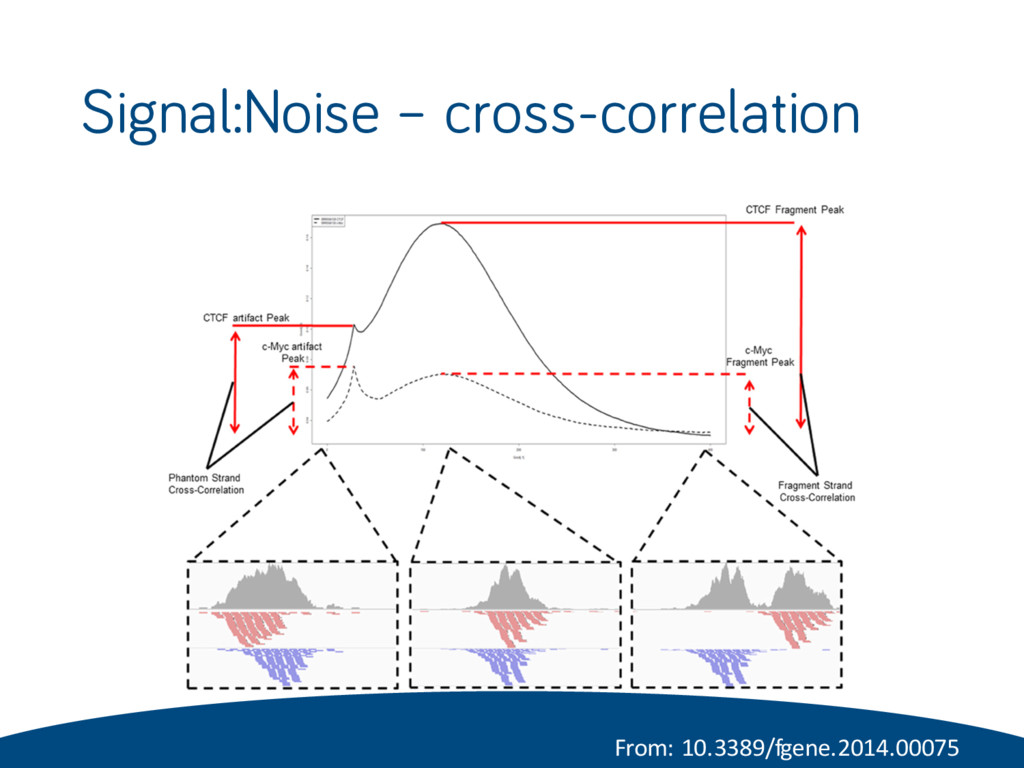
binding • Difficult to determine until after sequencing • Evaluated by number and strength of peaks: FRiP (fraction of reads in peaks): Npeak /Nnonred NSC (normalised strand coefficient) = Cfrag/ Cmin RSC (relative strand correlation) = (Cfrag − Cmin )/(Cread − Cmin ) • C = correlation (Pearson) between mapped read densities of positive and negative strands (y-axis) with shifting one strand (x- axis) • The ENCODE consortium recommends an NSC ≥ 1.05 and an RSC ≥ 0.8 for typical TFs (sharp mode)
are prepared from a small amount of starting materials • Measured by the non-redundant fraction (NRF) NRF = Nnonred /Nall • Nnonred = reads mapped to same genomic coordinates T times or fewer • In practice, T is usually 1 • The ENCODE consortium endorses an NRF > 0.8 for 10 million reads (T = 1)
step • Known as input • Other controls often requested by reviewers (so consider up front) • RNAi knock-down of IP target • Non-binding control (IgG, GFP) • Also note absolute requirement by some journals for n>=2 • Agreement between replicates another key QC test • Test with Jaccard statistic (raw reads) • Or correspondence of peak profiles
factors • Difficult to determine a priori • Can use post hoc saturation analysis • Systematically drop out data and look for plateau point of peaks detected • Especially for histone modifications, there doesn’t appear to be a practical saturation point • More reads == more peaks • RULES OF THUMB • For sharp peaks, 10m uniquely mappable reads per replicate • For broad peaks. 40-50m reads as ’practical minimum’
sample conditions and target proteins. Even though there is no clear consensus on which is best, the latest and widely used programs may be satisfactory for our needs.” From: 10/1093/bib/bbw023
Even for TF with canonical motif, condition- specific binding independent of motif can and does occur • The irreproducible discovery rate (IDR) assesses the rank consistency of common peaks between two replicates • One ’failed’ sample will become bottleneck • So bad replicates must be discarded • Makes experiments prone to batch effects • Poor quality samples basically unrecoverable
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}