Design and De Novo assembly using the PacBio® HGAP method After the training, you will be able to • Understand how the HGAP method works • Understand the coverage targets for de novo assembly with PacBio® data • Import and parameterize an HGAP assembly job in SMRT® Portal • SMRT® Technology • PacBio® System Workflow
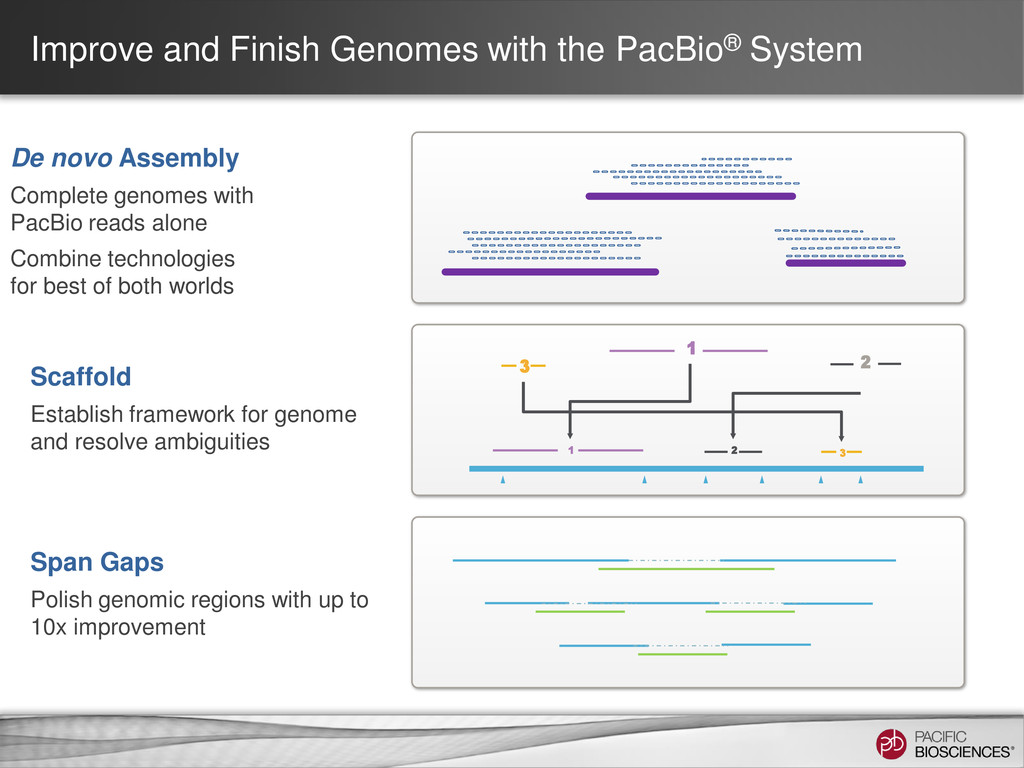
Assembly Complete genomes with PacBio reads alone Combine technologies for best of both worlds 2 3 2 3 1 1 Scaffold Establish framework for genome and resolve ambiguities Span Gaps Polish genomic regions with up to 10x improvement
Sequencing reads with short reads for error correction • Requires multiple types of data, (at least) two library preps, different sequencing technologies…
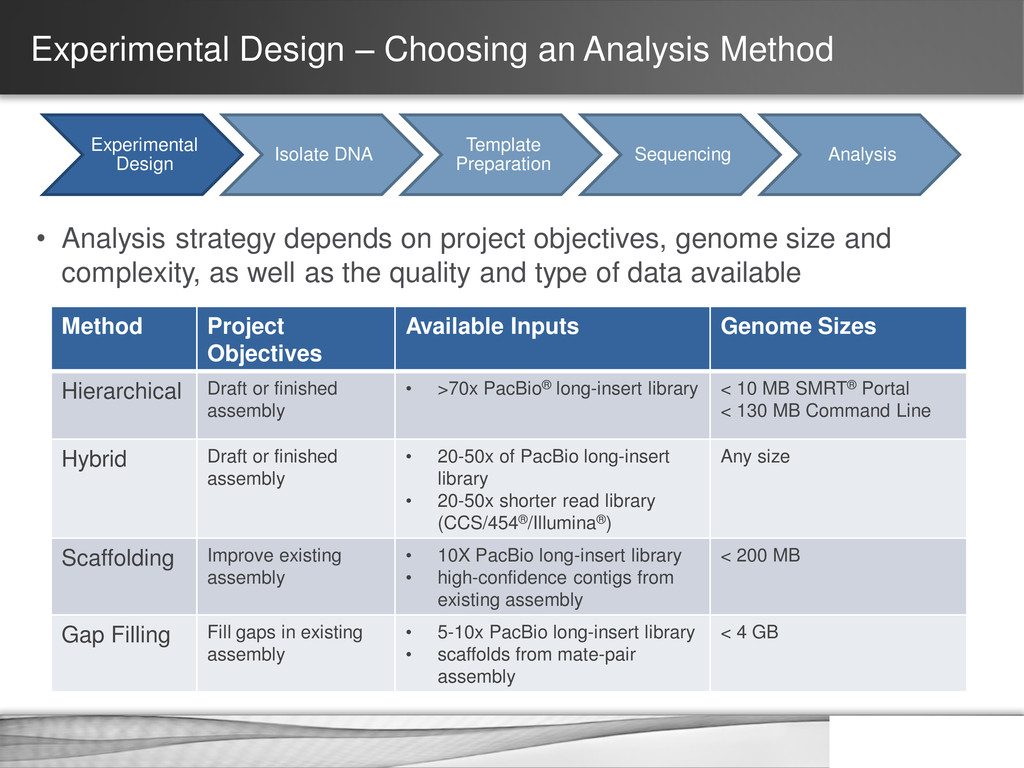
DNA Template Preparation Sequencing Analysis • Analysis strategy depends on project objectives, genome size and complexity, as well as the quality and type of data available Method Project Objectives Available Inputs Genome Sizes Hierarchical Draft or finished assembly • >70x PacBio® long-insert library < 10 MB SMRT® Portal < 130 MB Command Line Hybrid Draft or finished assembly • 20-50x of PacBio long-insert library • 20-50x shorter read library (CCS/454®/Illumina®) Any size Scaffolding Improve existing assembly • 10X PacBio long-insert library • high-confidence contigs from existing assembly < 200 MB Gap Filling Fill gaps in existing assembly • 5-10x PacBio long-insert library • scaffolds from mate-pair assembly < 4 GB
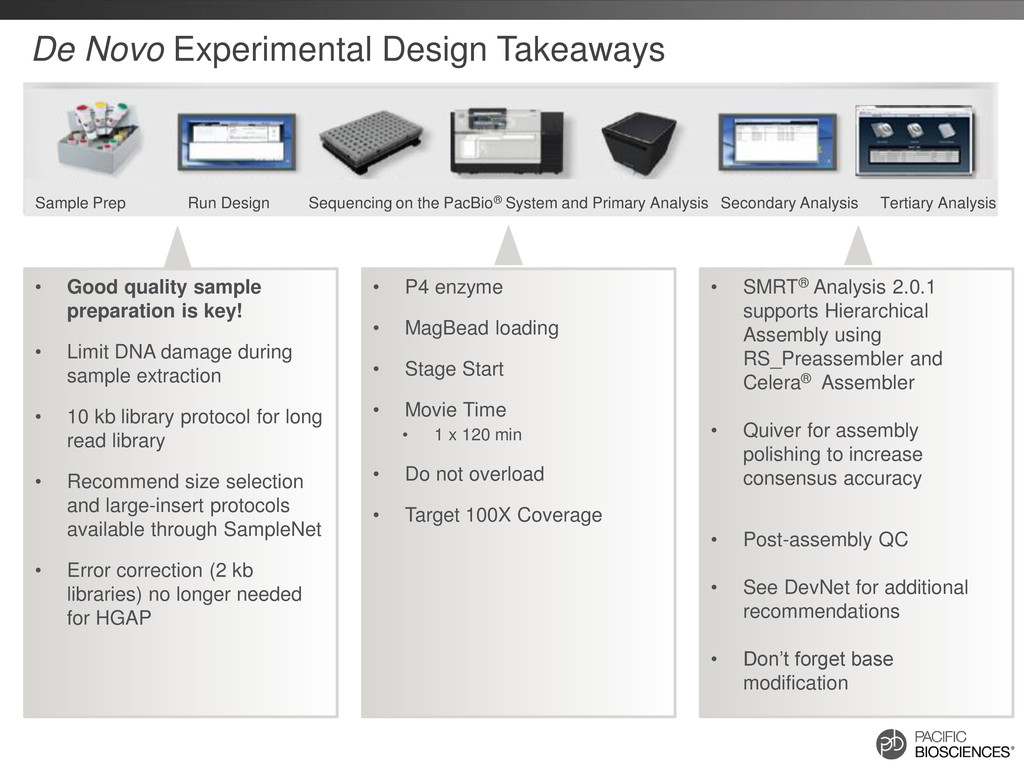
performance • No amplification step during library preparation • Recommendations: – Take care during extraction to avoid gDNA damage & avoid contaminants – Use extraction methods or kits that produce very high molecular weight gDNA – If contaminants are present, purify starting DNA material prior to library prep – Accurately quantify and qualitatively evaluate gDNA – Include DNA-damage-repair step in library prep 29 Experimental Design Isolate DNA Template Preparation Sequencing Analysis
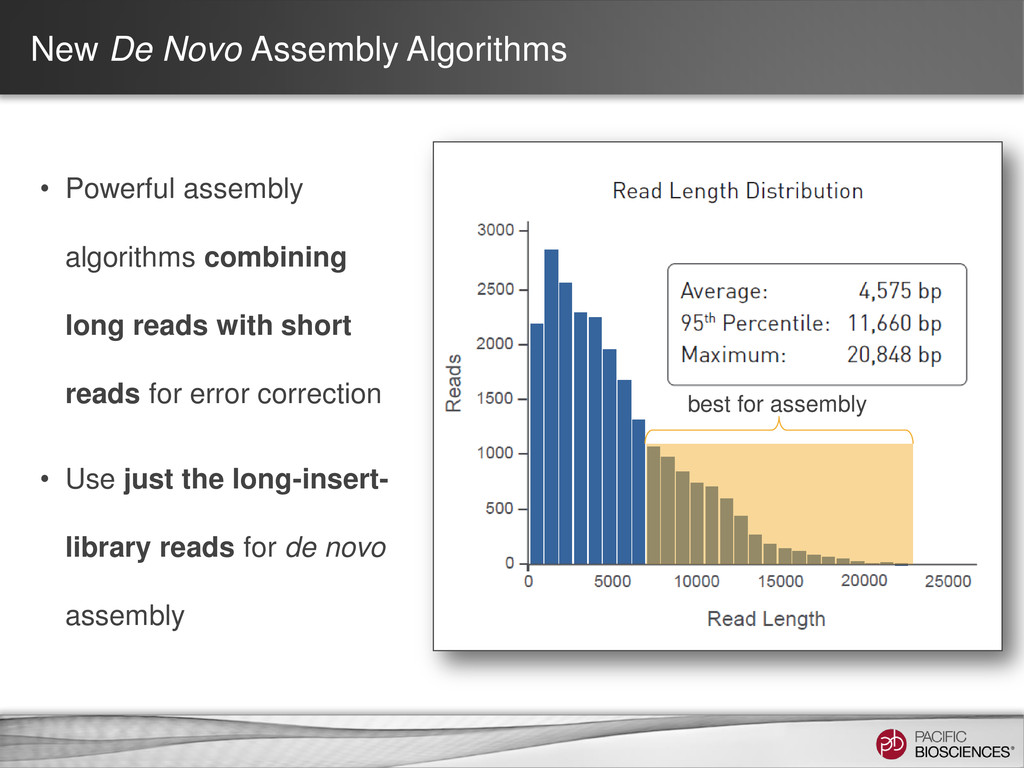
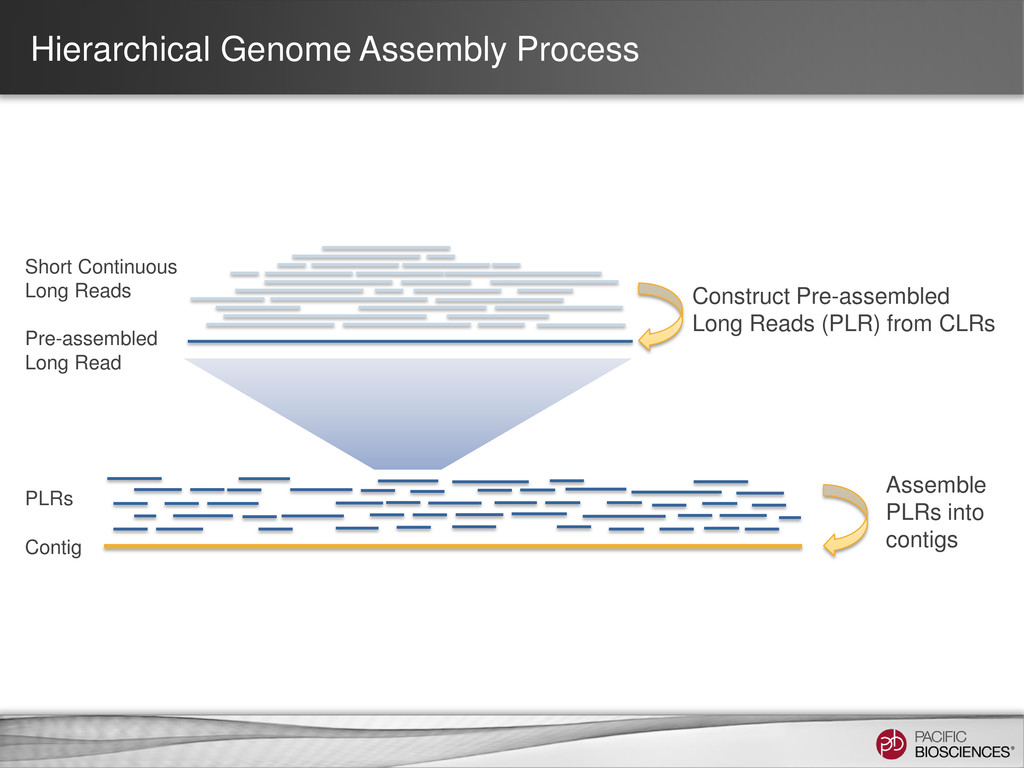
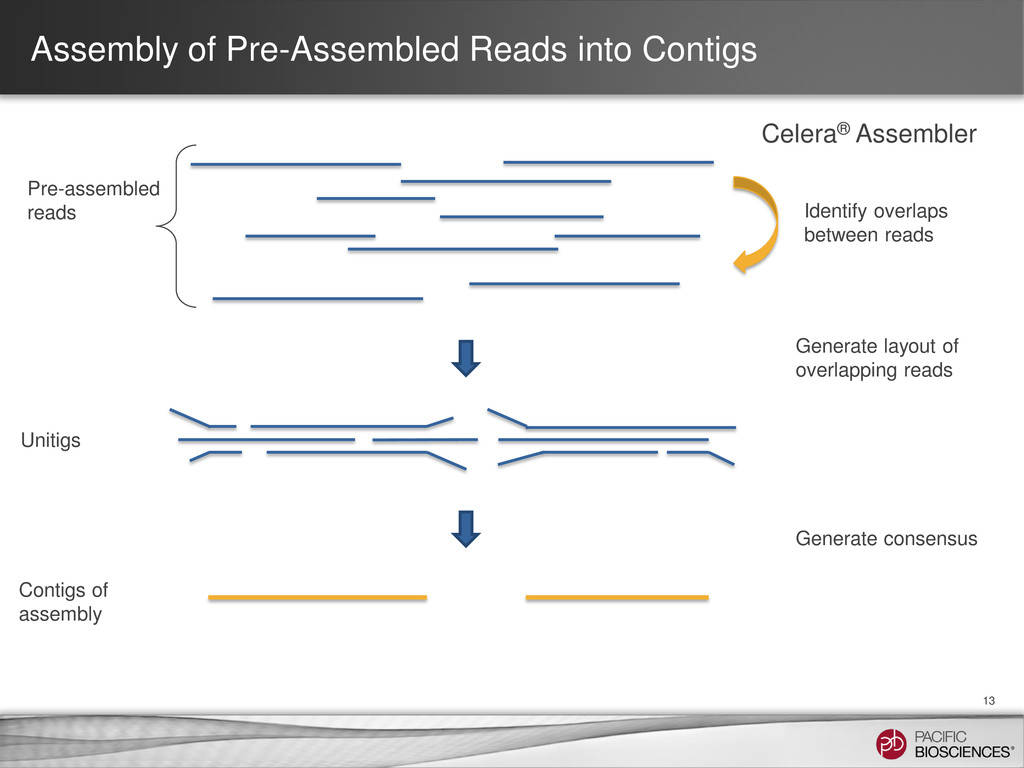
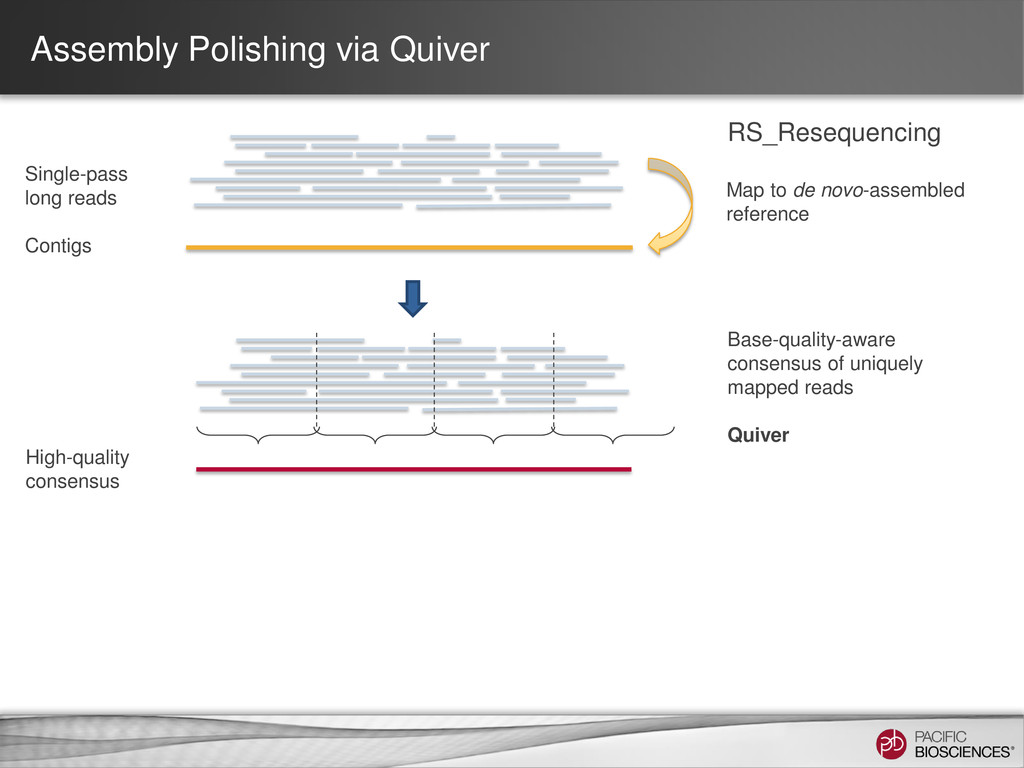
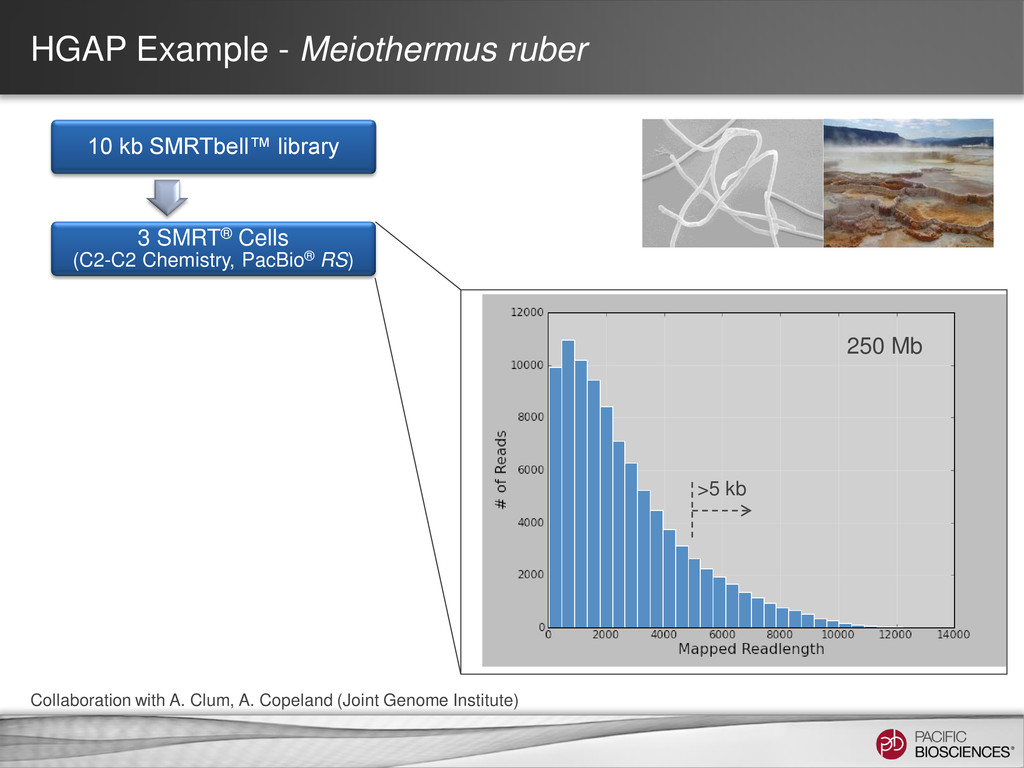
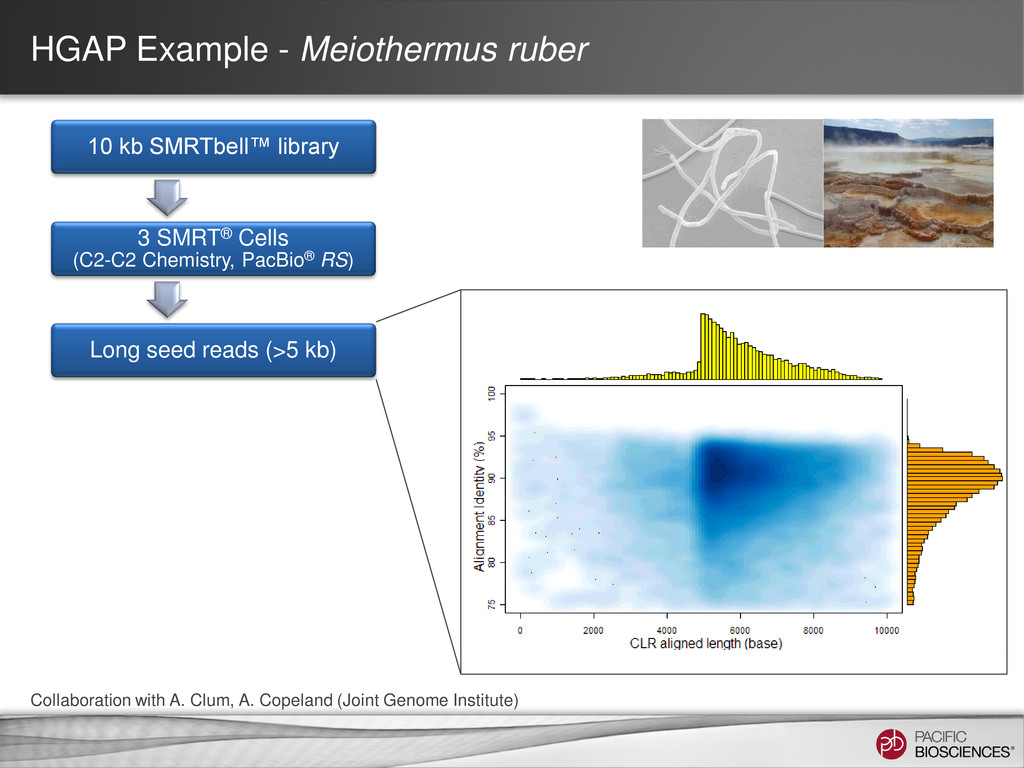
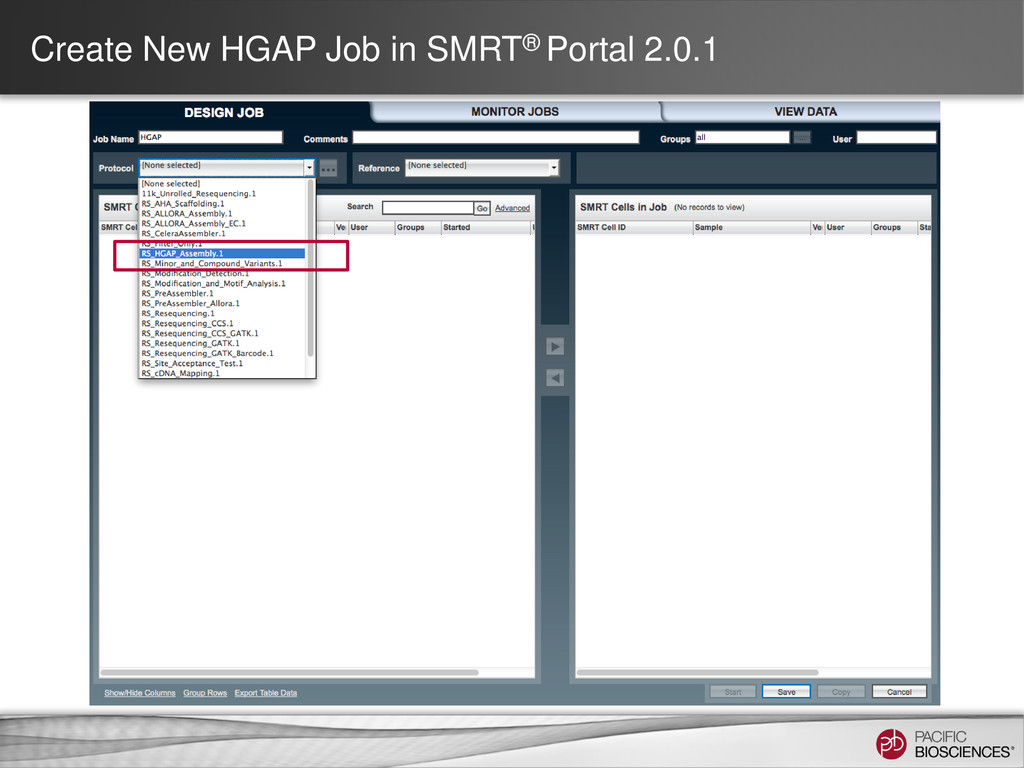
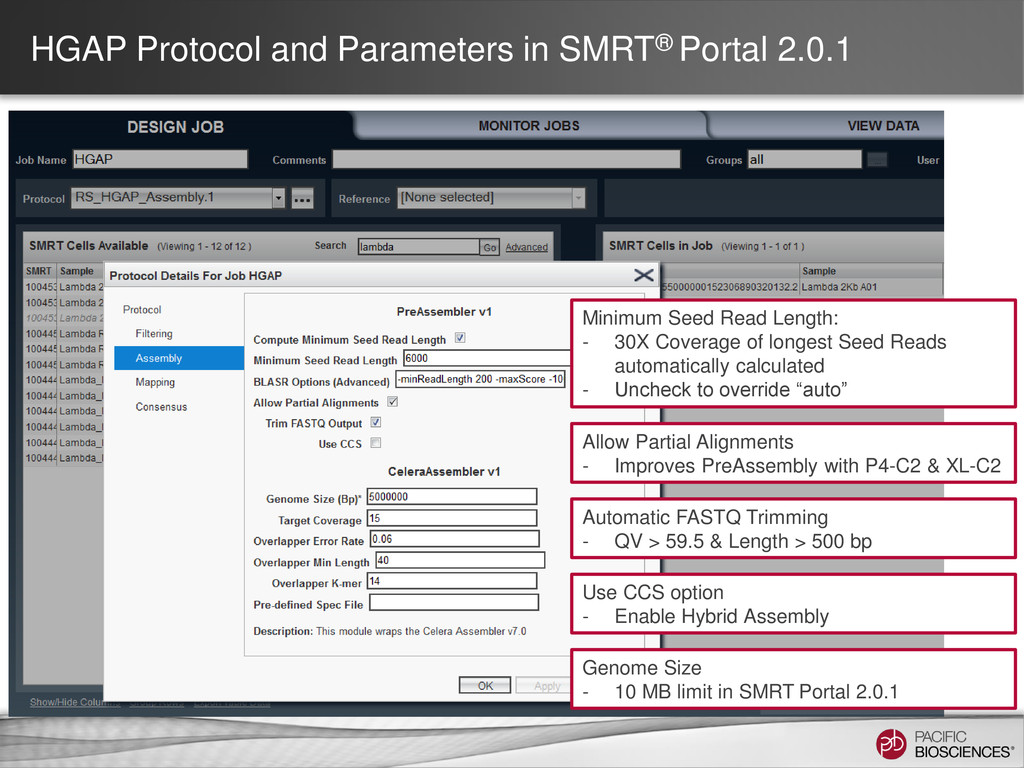
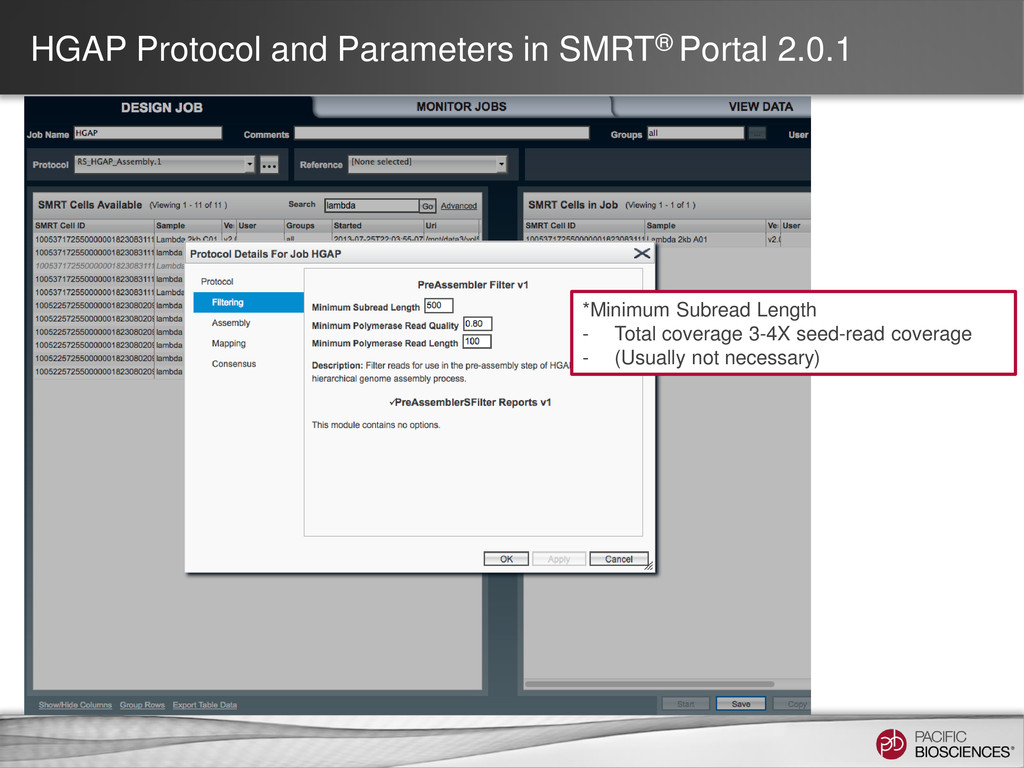
loading • Stage Start • Movie Time • 1 x 120 min • Do not overload • Target 100X Coverage • SMRT® Analysis 2.0.1 supports Hierarchical Assembly using RS_Preassembler and Celera® Assembler • Quiver for assembly polishing to increase consensus accuracy • Post-assembly QC • See DevNet for additional recommendations • Don’t forget base modification • Good quality sample preparation is key! • Limit DNA damage during sample extraction • 10 kb library protocol for long read library • Recommend size selection and large-insert protocols available through SampleNet • Error correction (2 kb libraries) no longer needed for HGAP Sample Prep Run Design Sequencing on the PacBio® System and Primary Analysis Secondary Analysis Tertiary Analysis
of presentations, posters and other de novo assembly resources available through PacBio’s website (www.pacb.com/denovo) • Protocols, Technical & Application Notes available through Customer Portal • DevNet – HGAP Reference Implementation: http://www.smrtcommunity.com/Share/Code?id=a1q70000000H2qRAAS – Quiver: www.pacbiodevnet.com/quiver – Bacterial Assembly and Epigenetic Analysis Training Web Video http://www.pacificbiosciences.com/Tutorials/Bacterial_Assembly_Epigenetic_Analysis_HGA P/story_html5.html • Additional information on Assembly Tools – Celera® Assembler: http://sourceforge.net/apps/mediawiki/wgs- assembler/index.php?title=PacBioToCA – Allpaths-LG: http://www.broadinstitute.org/software/allpaths-lg/blog/ – PBJelly: http://sourceforge.net/p/pb-jelly/wiki/Home/ 33
are trademarks of Pacific Biosciences in the United States and/or other countries. All other trademarks are the sole property of their respective owners.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}