London Ser. A 253, 242 (1959). [2] S. Huzinaga and A. A. Cantu, J. Chem. Phys. 55, 5543 (1971). [3] S. Huzinaga, D. McWilliams, and A. A. Cantu, Adv. Quantum Chem. 7, 187 (1973). [4] V. Lua˜ na, M. Bermejo, M. Fl´ orez, J. M. Recio, and L. Pueyo, J. Chem. Phys. 90, 6409 (1989). [5] V. Lua˜ na and L. Pueyo, Phys. Rev. B 39, 11093 (1989). [6] V. Lua˜ na and L. Pueyo, Phys. Rev. B 41, 3800 (1990). [7] E. Francisco, A. Mart´ ın Pend´ as, and W. H. Adams, J. Chem. Phys. 97, 6504 (1992). [8] A. M. Pend´ as, E. Francisco, and J. M. Recio, J. Chem. Phys. 97, 452 (1992). [9] V. Lua˜ na, J. M. Recio, and L. Pueyo, Phys. Rev. B 42, 1791 (1990). [10] M. A. Blanco, E. Francisco, and V. Lua˜ na, Comput. Phys. Commun. 158, 57 (2004), source code distributed by the CPC program library: http://cpc.cs.qub.ac.uk/summaries/ADSY. [11] J. M. Recio, A. M. Pend´ as, E. Francisco, M. Fl´ orez, and V. Lua˜ na, Phys. Rev. B 48, 5891 (1993). [12] A. M. Pend´ as, V. Lua˜ na, J. M. Recio, M. Fl´ orez, E. Francisco, M. A. Blanco, and L. N. Kantorovich, Phys. Rev. B 49, 3066 (1994). [13] M. ´ Alvarez Blanco, Tesis doctoral, Universidad de Oviedo, 1997, director: V´ ıctor Lua˜ na Cabal. [14] M. A. Blanco, A. Costales, A. M. Pend´ as, and V. Lua˜ na, Phys. Rev. B. 62, 12028 (2000). [15] M. Fl´ orez, J. M. Recio, E. Francisco, M. A. Blanco, and A. M. Pend´ as, Phys. Rev. B 66, 144112/1 (2002). [16] V. Lua˜ na, J. M. Recio, A. M. Pend´ as, M. A. Blanco, L. Pueyo, and R. Pandey, Phys. Rev. B 64, 104102 (2001). [17] M. A. Blanco, A. Costales, V. Lua˜ na, and R. Pandey, Appl. Phys. Lett. 85, 4376 (2004). [18] R. F. W. Bader, Atoms in Molecules. A Quantum Theory (Oxford University Press, Oxford, 1990). [19] P. Mori-S´ anchez, Tesis doctoral, Universidad de Oviedo, 2002, directores: V´ ıctor Lua˜ na Cabal y Angel Mart´ ın Pend´ as. [20] V. Lua˜ na, A. Costales, P. Mori-S´ anchez, M. A. Blanco, and A. Mart´ ın Pend´ as, Acta Cryst. A. 60, 434 (2004). [21] P. Mori-S´ anchez, A. Mart´ ın Pend´ as, and V. Lua˜ na, J. Am. Chem. Soc. 124, 14721 (2002). [22] V. Lua˜ na, P. Mori-S´ anchez, A. Costales, M. A. Blanco, and A. Mart´ ın Pend´ as, J. Chem. Phys. 119, 6341 (2003). [23] A. Costales, M. A. Blanco, P. Mori-S´ anchez, V. Lua˜ na, and A. Mart´ ın Pend´ as, J. Phys. Chem. A 108, 2794 (2004). [24] A. M. Pend´ as, A. Costales, M. A. Blanco, J. M. Recio, and V. Lua˜ na, Phys. Rev. B. 62, 13970 (2000). c V. Lua˜ na 2005 (35)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
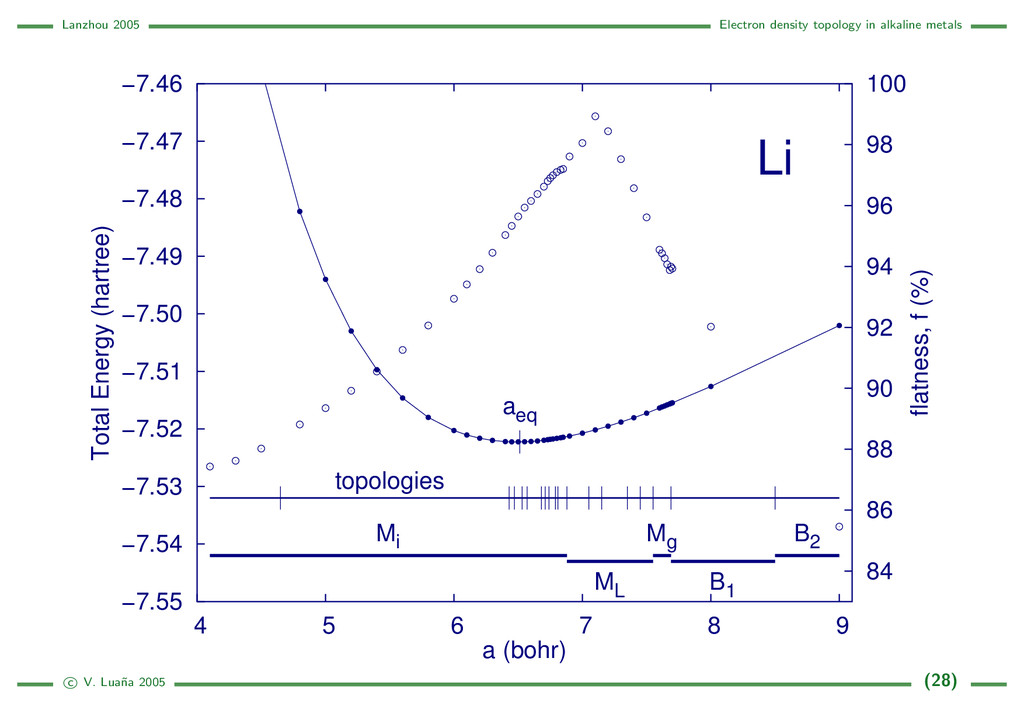{kind=link}
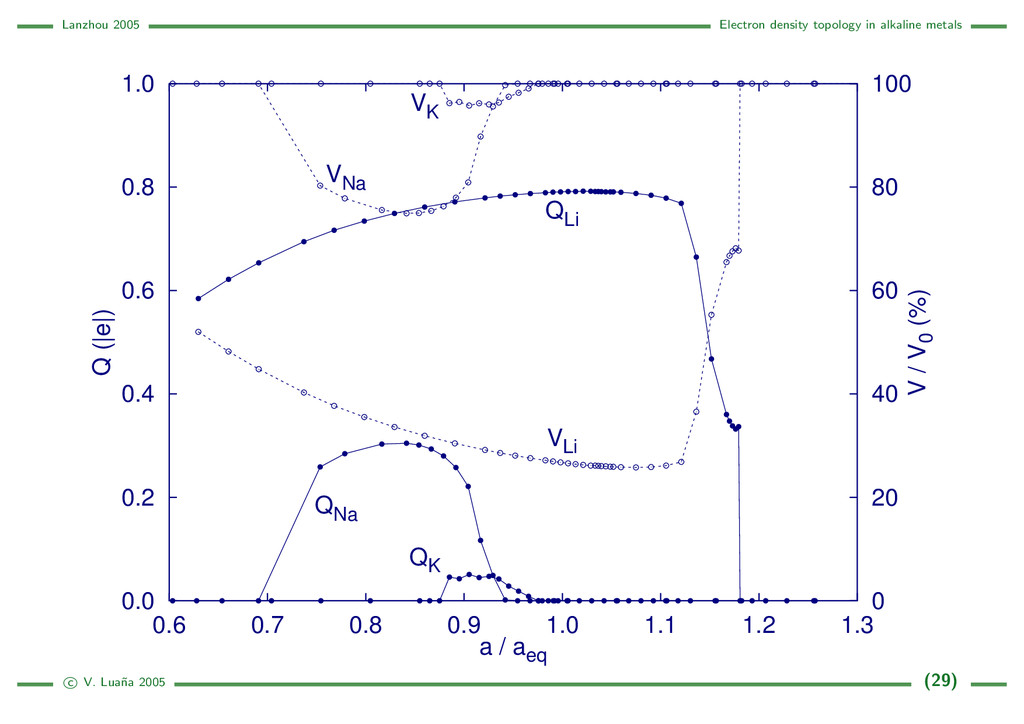{kind=link}
{kind=link}
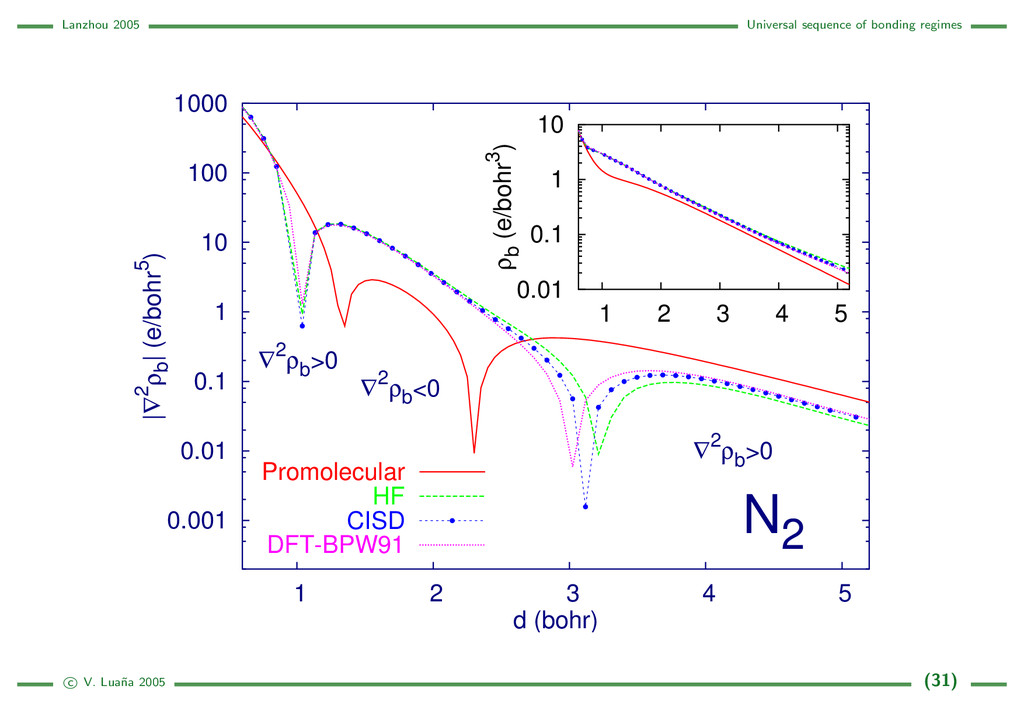{kind=link}
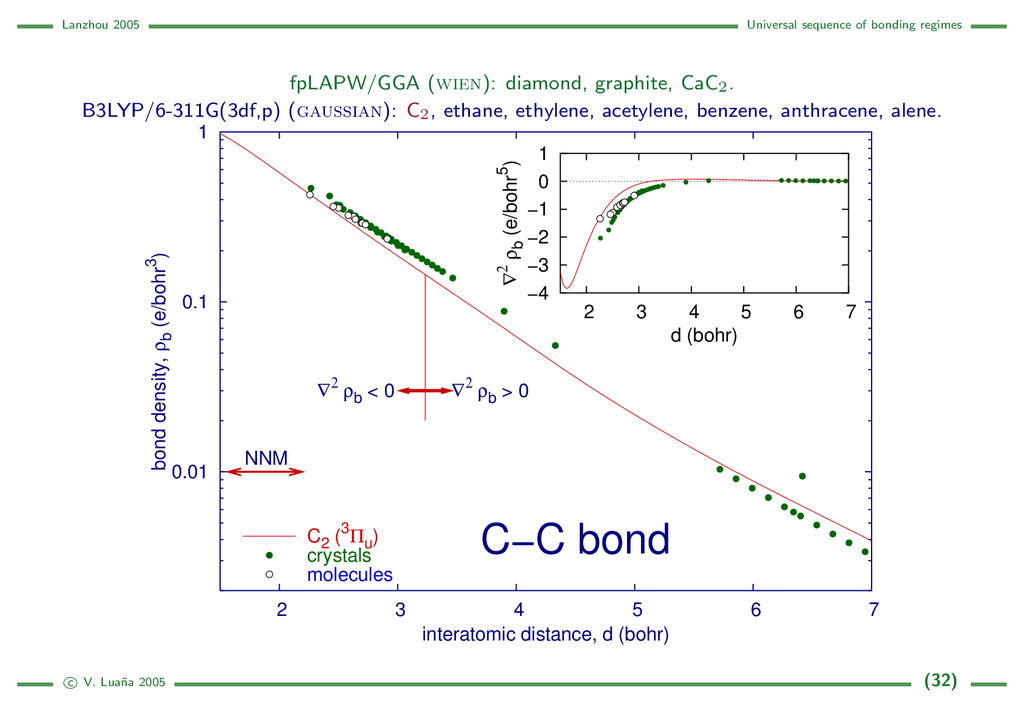{kind=link}
{kind=link}
{kind=link}
![Lanzhou 2005 References References [1] R. McWeeny, Proc. R. Soc.](https://files.speakerdeck.com/presentations/4c78d1b069b9013170b6769b07168819/slide_34.jpg){kind=link}